ChIP-seq эмбрионов дрозофилы
Протокол ChIP-seq 12 часовых эмбрионов дрозофилы.
Данный протокол был опробован для эксперимента ChIP-seq с замороженными в PBS 12-часовыми эмбрионами дрозофилы. В качестве антител использовали anti-H3K27me3 от Abclonal (A22006). Обратите внимание, что дехорионизация и удаление вителлиновой оболочки в данном протоколе не делается, поскольку на данной стадии число эмбриональных ядер очень велико. Кроме того, здесь не обсуждается процедура сбора эмбрионов. В разделе “Источники” можно найти литературу по этому поводу.
Процедура ChIP включает несколько этапов. На первом этапе (первый день) образец отмывается, фиксируются формальдегидом и гомогенизируются. После фиксации клеточные ядра отмываются буферами для отмывки, затем ресуспендирются в буфере для соникирования и подвергаются обработке ультразвуком. После фрагментации нужное количество хроматина смешивается с антителами на магнитных шарах в буфере для иммунопреципитации. После 16-часового осаждения на антителах (второй день), хроматин отмывается буферами с постепенным увеличением ионной силы. На последнем этапе осажденный хроматин ресуспендируется в буфере для элюции, обрабатывается протеиназой и РНКазой и декросслинкируется 16 часов. Затем ДНК очищается фенол-хлороформным методом (третий день).
Продолжительность ChIP от начала фиксации до выделения ДНК занимает 3 дня. Причем остановить эксперимент возможно только в первый день. Поэтому необходимо предварительно с особой внимательностью распределить рабочее время. Второй день эксперимента следует поставить не позже четверга, иначе придется работать в выходные.
Чеклист
Перед началом эксперимента следует проверить наличие всех инструментов, реактивов и расходников из списка.
Буферные растворы
Растворы готовятся непосредственно перед экспериментом, чтобы избежать зароста.
Все растворы нужно готовить с запасом не менее 10%.
Перед использованием к большинству растворов примешивается рабочая концентрация протеазных ингибиторов. Этого можно не делать в случае с контрольными экспериментами, когда проверяется только фрагментация или выход ДНК в Input. Состав ингибиторов может сильно варьировать в зависимости от типа материлов и вида антител. Например, при работе с ТФ и меткой H3K27ac важно добавлять к растворам бутират, ингибирующий деацетилазы гистонов. В данном протоколе все рецепты написаны с указанием DTT, PMSF и коомерческого коктейля ингибиторов PIC. Последний достаточно доргой, поэтому добавляется только на тех этапах, где продолжительность действия протеаз на хроматин больше 1 часа. Важно понимать, что добавление любого из ингибиторов опционально и может вообще не повлиять на выход ДНК.
1 день
Фиксация замороженных эмбрионов
- 15-50 мкл замороженных эмбрионов разморозить и отмыть в PBS. Осадить центрифугированием 2000g 5 минут, КТ. Удалить супернатант полностью.
- Отмыть эмбрионы в 1 мл буфера А1 комнатной температуры. Осадить центрифугированием 2000g 5 минут, КТ.
- Ресуспендировать эмбрионы в 1 мл буфера А1 комнатной температуры. Перенести 973 мкл эмбрионов в гомогенизатор Dounce (2 мл). Добавить 27.7 мкл 37% формальдегида. Начать отсчитывать время фиксации. В течение четырех минут растирать пестиком (~15 медленных движений без пузырения).
Рабочая концентрация формальдегида - 1%.
- Сразу перенести гомогенизат в эппендорф и поставить на мешалку (КТ). По окончании 10 минут фиксации поставить добавить 120 мкл 2.5 М холодного глицина (финальная концентрация глицина получится 250 мМ), перемешать и поставить в ледна пять минут.
Перед добавлением глицина нужно проверить объем фиксируемого материала. Если меньше 1 мл, то объем глицина нужно уменьшить пропорционально.
- Центрифугировать 2-5 минут при 2000g 4оС. Удалить супернатант, не задевая осадок.
- Промыть осадок пипетированием в 1 мл холодного буфера А1. Снова центрифугировать 2-5 минут при 2000g 4оС.
- Повторить отмывку холодным буфером А1. Снова центрифугировать 2-5 минут при 2000g 4оС. Удалить супернатант, не задевая осадок.
Осадок можно заморозить в кельвинаторе. После отмывки его можно фрагментировать. Это снизит выход ChIP.
- Пипетировать осадок в 1 мл Wash Solution I + ингибиторы. Инкубировать во льду 10 минут при постоянном перемешивании. Центрифугировать 2-5 минут при 2000g 4оС. Удалить супернатант, не задевая осадок.
- Пипетировать осадок в 1 мл Wash Solution II + ингибиторы. Инкубировать во льду 5 минут при постоянном перемешивании. Центрифугировать 2-5 минут при 2000g 4оС. Удалить супернатант полностью, не задевая осадок.
- Аккуратно промыть осадок в 1 мл Sonication buffer. Буфер наносить по стенке, чтобы не повредить осадок. Сразу удалить буффер, не задевая осадок.
Осадок можно заморозить в кельвинаторе. Но это с большой вероятностью несколько снизит эффективнось иммунопреципитации.
Далее хроматин подвергается соникированию. Если иммунопреципитация ставится в тот же день, то нужно заранее поставить подготовку антител (пункты 11-14). Нужно учитывать, что суммарно это занимает не менее 2 часов.
Подготовка антител (+ преблокирование, опционально).
При работе с шарами MagnacChIP A+G нужно избегать интенсивного встряхивания на шейкере.
Если блокировать антитела не обязательно, то все этапы проводят с PBS+0.5% Tween. В противном случае необходимо использовать Blocking Buffer. Приготовить выбранный буфер из расчета 1.5 мл на аликвоту шариков. 1 мл поставить в лед, остальное довести до комнатной температуры.
Как показал опыт, при КТ антитела сохраняют активность вплоть до 6 часов. Поэтому нет ничего страшного, если процедура замешивания антител с шариками затянется более 2 часов.
- Отобрать по 10-20 мкл MagnaСhIP A+G на AB+ и AB- в отдельные эппендорфы. Промыть в 500 мкл PBS+0.5% Tween или Blocking Buffer комнатной температуры. Перемешивать 5 минут при КТ на роторной мешалке. Поставить на 1 минуту на магнитный штатив и удалить супернатант.
Магнитные частицы имеют свойство слипаться друг с другом и со стенками пробирок, если в растворе отсутствует детергент. По этой причине для отмывки шаров в раствор обязательно нужно добавить Tween-20.
При вращении мешалки жидкость в пробирке должна обязательно переливаться от дна к крышке, иначе шарики осядут на стенки. Tween необходим, чтобы шарики не слипались.
- Повторить отмывку в 500 мкл PBS+0.5% Tween или Blocking Buffer комнатной температуры. Перемешивать 5 минут при КТ на роторной мешалке. Поставить на 1 минуту на магнитный штатив и удалить супернатант.
- Ресуспендировать шары в 200 мкл мкл PBS+0.5% Tween или Blocking Buffer комнатной температуры. Добавить выбранное количество антител (обычно мы используем 1 мкг) к пробирке для AB+. К AB- антитела не добавлять! Поставить на роторную мешалку на 2 часа при КТ.
- Этот шаг делается непосредственно перед иммунопреципитацией, когда хроматин будет разведен в Immunoprecipitation buffer (пункт 20)! Дважды отмыть шарики с присоединенными антителамив 500 мклледяного PBS+0.5% Tween или Blocking Buffer аналогично пункту 11. После окончательной отмывки удалить супернатант полностью. Незамедлительно приступить к иммунопреципитации.
Фрагментация хроматина
- Ресуспендировать осадок в ледяном Sonication buffer + PIC из расчета ~1 мкг хроматина на 100 мкл (300 мкл на 25-50 мкл эмбрионов).
- Расфасовать по 100 мкл в 600мкл пробирки для соникирования. Осадить, чтобы капли жидкости не остались на стенках и поставить в лед для максимального охлаждения.
- Фрагментировать в режиме 30 cек. ON - 30 сек OFF в течение 16 циклов.
При озвучивании жидкость разбрызгивается по стенкам пробирки, поэтому процесс соникирования стоит разделить на два этапа, чтобы в промежутке между ними осадить капли со стенок пробирки при помощи центрифуги.
В результате соникирования размер фрагментов ДНК должен достичь промежутка 100-1000 пн с медианой в области 200-300 пар. Допускается небольшой хвост из фрагментов длинее 1 кб. Фрагментный состав итогового продукта зависит от продолжительности соникирования и степени фиксации. В данном протоколе фиксация проводится в 1% формальдегиде в течении 10 минут при КТ. По опыту, для достижения необходимого фрагментного состава при таких вводных потребуется где-то 16-20 циклов в режиме 30'‘ON - 30'‘OFF. В общем случае, продолжительность фрагментации определяется заранее в предварительном эксперименте.
- Объединить озвученный хроматин в эппендорфе. Центрифугировать на максимальных оборотах 5 минут при 4оС. Перенести супернатант в чистую пробирку. Поставить в лед. Тщательно измерить объем полученного хроматина. Довести до нужного объема добавлением ледяного Sonication buffer + PIC.
Озвученный хроматин можно заморозить в кельвинаторе. Но это несколько снизит выход ChIP.
- Рассчитать объем хроматина для AB+, AB- и Input. В инпут подбирается объем, достаточный для выделения 100-500 нг ДНК. На 300 мкл соникированного хроматина достаточно взять 14 мкл на Input, 140 мкл на AB+, 140 мкл на AB-.
Судя по рекоммендациям из протоколов ChIP-seq, максимально точно определить концентрацию ДНК в фрагментированном хроматине можно с помощью Qubit. Однако обычные прикидочные подсчеты, как правило, работают очень хорошо.
Объем AB + и AB - должны быть равны. Разделять аликвоты AB + и AB - нужно будет только после пункта 21.
- Записать долю объема Input от AB+ в процентах. Заморозить Input в морозильной камере.
- Довести состав раствора с хроматином в AB+ и AB- до Immunoprecipitation buffer (150 mM NaCl)+PIC, добавив соответствующий объем ледяного preImmunoprecipitation Buffer. Для приготовления последнего нужно знать объем AB+ плюс AB- (X) и суммарный объем, в котором будет проходить реакция с шариками и хроматином (Y). К X мкл хроматина в Sonication Buffer нужно добавить Y - X мкл preImmunoprecipitation Buffer. Чтобы прописать состав preImmunoprecipitation Buffer мы учитываем, что некоторые компоненты уже присутствуют в Sonication Buffer.
Удобно проводить иммунопреципитацию в объеме 300 мкл. Так что нужно довести суммарный объем фрагментированного хроматина до Y=600 мкл. Если фрагментация проводилась в объеме 300 мкл, то после извлечения Input (14 мкл) удобно, чтобы объем хроматина для AB+ составлял 140 мкл, то есть всего мы возьмем 280 мкл на AB+ и AB- (X=280).
preImmunoprecipitation Buffer
| Компонент | Стоковая конц. | Рабочая конц. | Целевой объем, мкл | Сколько добавить, мкл | Y=600 мкл X=280 мкл Y-X=320 мкл |
|---|---|---|---|---|---|
| HEPES, pH8.0 | 1 М | 50 мМ | Y | 50*Y/1000 | 30 мкл |
| EDTA | 0.5 М | 1 мМ | Y-X | 1*(Y-X)/500 | 0.64 мкл |
| NaCl | 5 M | 150 мМ | Y | 150*Y/5000 | 18 мкл |
| NaDOC | 10% | 0.1% | Y | 0.1*Y/10 | 6 мкл |
| SDS | 10% | 0.1% | Y-X | 0.1*(Y-X)/10 | 3.2 мкл |
| Triton-X100 | 100% | 1% | Y | 1*Y/100 | 6 мкл |
| DTT | 1 М | 0.5 мМ | Y-X | 0.5*(Y-X)/1000 | 0.16 мкл |
| PMSF | 1 М | 1 мМ | Y-X | 1*(Y-X)/1000 | 0.32 мкл |
| PIC | 7x | 1x | Y-X | 1*(Y-X)/7 | 45.1 мкл |
| mQ | Довести до Y-X | 210.6 мкл |
preImmunoprecipitation buffer готовится с запасом! Чтобы сделать запас 10 процентов, нужно все вычисленные объемы умножить на 1.1
- К этому моменту должны быть отмыты антитела на магнитных шариках (пункт 13). Перенести сначала половину фрагментированного хроматина в Immunoprecipitation buffer к шарам с антителами (AB+), затем оставшуюся часть к шарам без антител (AB-). Перемешать хроматин и шарики перемешиванием. Поставить пробирки на роторную мешалку и оставить вращаться в холодильнике или в холодной комнате (при +4оС) на 16 часов.
Обязательно проследить, чтобы пробирки не выпали из мешалки, и чтобы при вращении жидкость в каждой пробирке переливалась со дна на крышку.
День 2
Отмывка хроматина, лизис + декросслинкирование
- Промыть шары дважды в 500 мкл ледяного Immunoprecipitation buffer (150 mM NaCl ).
Для отмывок пробирки с шариками скручиваются, переносятся на магнитный штатив на минуту, после чего супернатант удаляется и заменяется буфером для отмывки. После замены буфера пробирки возвращяются на роторную мешалку на 5 минут при 4 градусах.
- Промыть дважды в 500 мкл ледяного Immunoprecipitation buffer c 300 мМ NaCl.
- Промыть дважды в 500 мкл ледяного DOC buffer.
- Промыть дважды в 1 мл ледяного TE.
Из-за отсутствия детергентов в ТЕ, магнитные шарики будут прилипать к стенкам пробирок. Поэтому нужно аккуратно удалять супернатант, чтобы не захватить шарики.
- Ресуспендировать шары в 300 мкл Elution buffer комнатной температуры**.**
- Разморозить Input и довести до 300 мкл с помощью Elution buffer комнатной температуры.
Дальнейшие реакции проводятся без охлаждения.
Далее Input , AB+ и AB- обрабатываются параллельно.
- Добавить 2-6 мкл РНКазы 10 мг/мл. Инкубировать 30 минут при 37 градусах в термошейкере.
- Добавить 12 мкл 1 M TrisHCl pH 8.0 , 6 мкл 0.5 М EDTA pH 8.0 и 14 мкл 5М NaCl.
Итоговая концентрация NaCl ~200 мМ. Соль нужна для последующего переосаждения ДНК спиртом.
- Добавить 2.5 мкл протеиназы К 20 мг/мл. Инкубировать в термошейкере 3 часа при 55оС (лизис белков), затем 16 часов при 64оС (декросслинкирование).
После завершения программы образец можно охладить до 4-10 градусов, однако это приведет к выпадению SDS в осадок. Растворить SDS можно нагреванием до 37 по Цельсию
День 3
Очистка ДНК
-
Лизат с магнитными частицами (AB+ и AB-) поместить на магнитный штатив на 1 минуту. Полностью перенести лизат в новую пробирку. Не забыть правильно подписать.
-
Добавить к лизатам равный объем фенол:хлороформа. Перемешать до образования эмульсии. Центрифугировать 5-10 минут при 13000g на КТ.
-
Перенести верхнюю фазу в чистый эппендорф. Добавить равный объем хлороформа. Аккуратно перемешать переворачиванием 10-13 раз. Центрифугировать 5-10 минут при 13000g на КТ.
Хлороформ обладает сильной текучестью, поэтому чтобы он не вытекал из носика под действием силы тяжести, чистый носик нужно смочить в хлороформе пипетированием.
- Перенести верхнюю водную фазу в чистую пробирку. Добавить 1.5 объема изопропанола. Добавить 1 мкл гликогена 20 мг/мл. Перемешать и поместить в морозильную камеру или кельвинатор на 20-30 минут. Центрифугировать 20 минут при 13000 g в центрифуге с охлаждением.
Обратить внимание на появление осадка. Если осадок не появился, значит нужно добавить гликоген еще раз и снова центрифугировать.
- Не задевая осадок, удалить супернатант.
Лучше не выливать супернатант, а перенести в чистый эппендорф на случай, если выход ДНК в AB+ и Input неожиданно окажется нулевым.
- Промыть осадок 70% этанолом. Центрифугировать 5 минут при 13000g в центрифуге с охлаждением. Полностью удалить этанол, не задевая осадок.
Если не удалось удалить этанол со стенок, нужно центрифугировать пробирку повторно и попытаться снова.
- Высушить осадок от спирта в течение 5 минут при КТ.
- Ресуспендировать осадок в 20 мкл mQ или EB.
Лучше не использовать mQ, который был расфасован и заморожен. Из-за сдвига pH продолжительность хранения ДНК в такой воде значительно снижается.
- ДНК можно заморозить !!!
Контроль качества
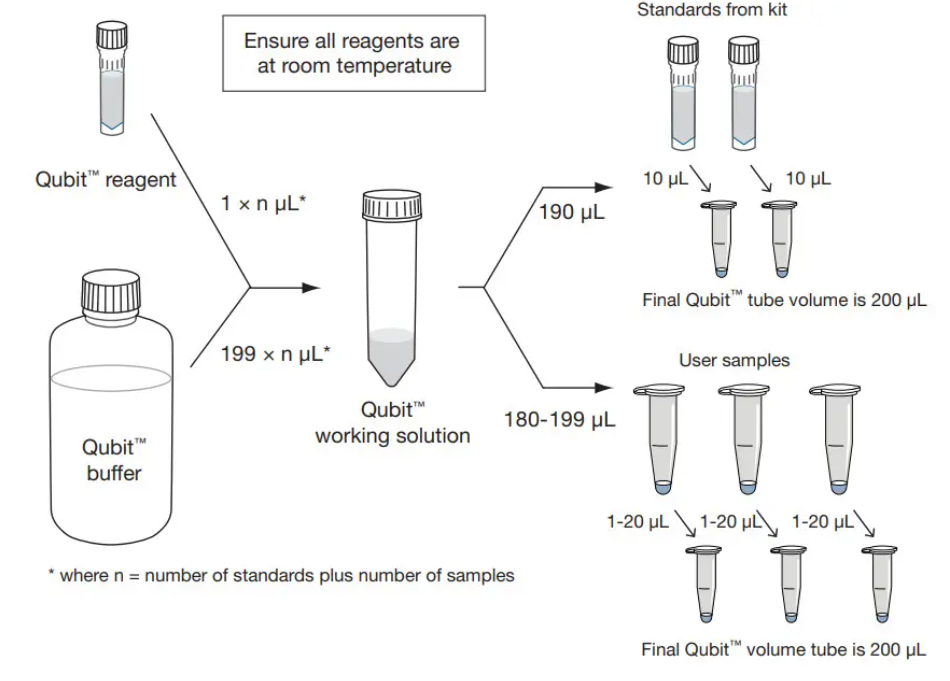
- Измерить концентрацию ДНК в AB+, AB- и Input при помощи Qubit с использованием набора реагентов dsDNA HS согласно схеме ниже.
На каждый образец необходимо приготовить 200 мкл 1xTE + 1xDye. Далее 199 мкл данного раствора перенести в специальную пробирку для Qubit и добавить 1 мкл образца.
При необходимости приготовить разведения стандартов (всего два стандарта: 0 нг/мкл и 10 нг/мкл. Для этого 10 мкл каждого стандарта смешивается с 190 мкл 1xTE + 1xDye.
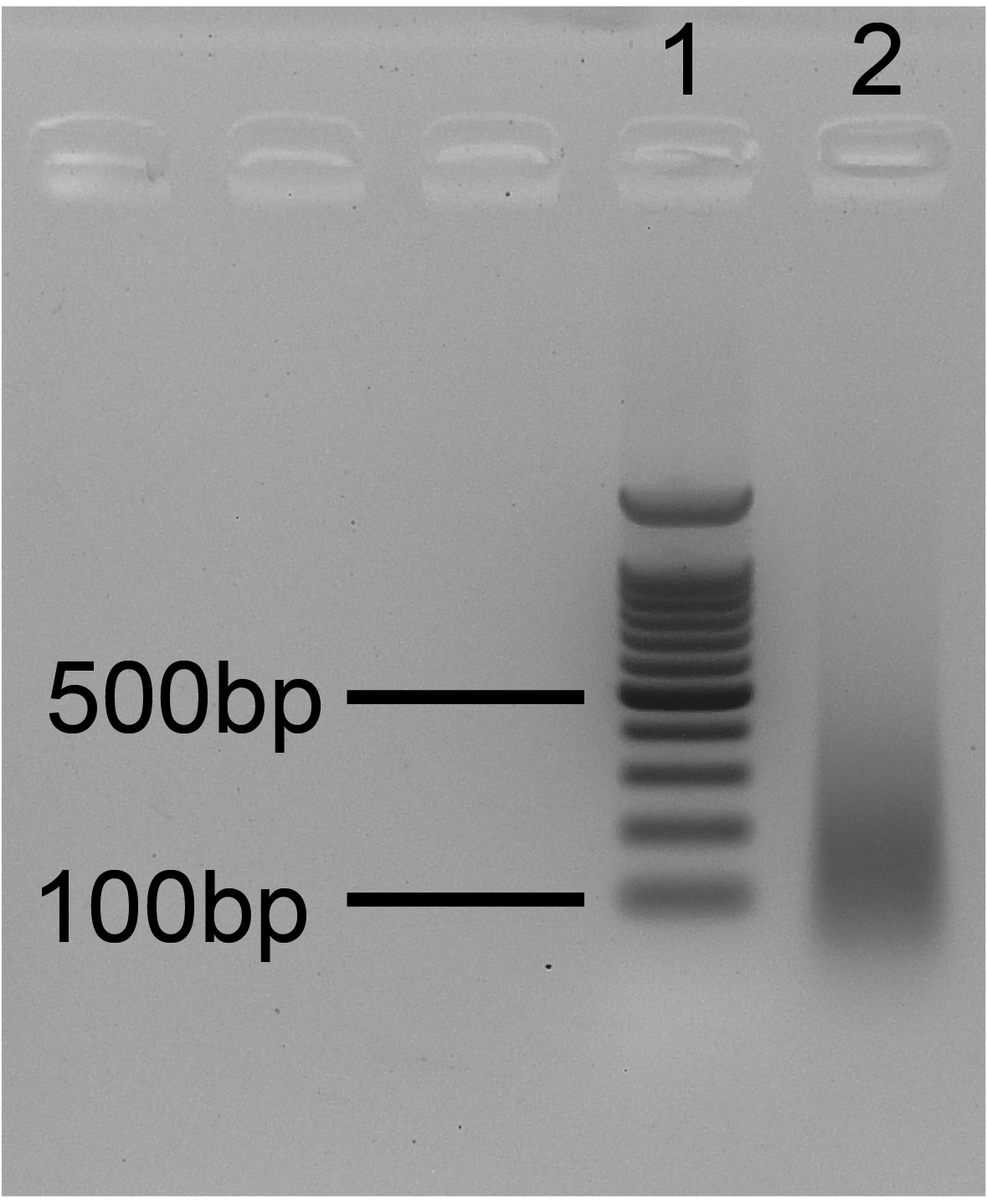
- Провести фрагментный анализ ДНК в Input при помощи электрофореза в 1.5% TAE-агарозе, 20-25 минут, 10 В/см, длина геля – 10 см (две стандартных дорожки). Этидий добавлять в гель перед заливкой. На форез необходимо добавить 50-150 нг ДНК. После нанесения на дорожку в образце Input должно остаться не менее 100 нг ДНК! В противном случае форез не ставить! Идеальное распределение по длине показано на диаграммах ниже.
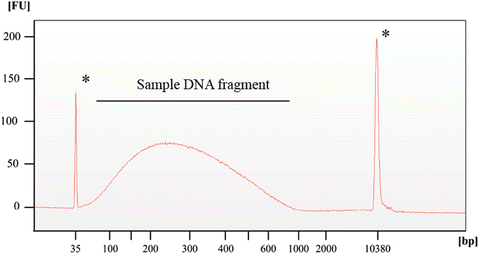
- C каждым полученным образцом провести qPCR для идентификации уровня обогащения в “+” и “–”-локусах (positive и negative control, соответственно). Степень обогащения оценивать по сравнению с Input. Для этого нужно измерить по каждому локусу отношение начальных концентраций ДНК в AB+ и AB- к Input при помощи формулы: X*CAB+/CInput и X*CAB-/CInput, где X – это доля Input по отношению к объему AB+ или AB- (образец IP и Mock, соответсвенно). Пример идеального эксперимента показан на диаграммах ниже:
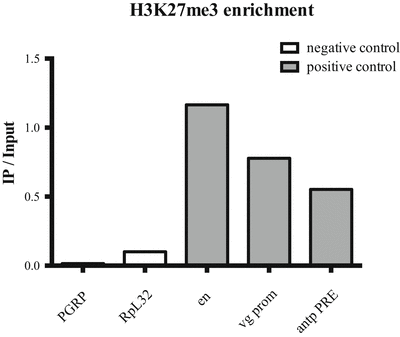
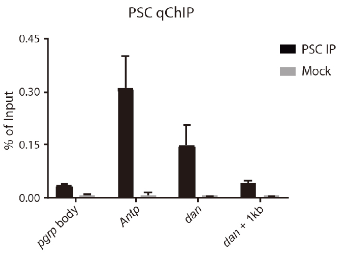
Во время всех проверок объем ДНК будет постепенно уменьшаться. Для успешной пробоподготовки библиотек с последующим секвенированием количество ДНК в AB+ и Input должно составлять не менее 10 нг. В идеале >100нг. Это должен быть неприкосновенный запас!
F.A.Q.
- Какой выход ожидается в AB+?
Заранее без предварительных экспериментов предсказать сложно, но можно оценить это. Если речь о ТФ, то нужно посчитать, сколько у него сайтов связывания и умножить на 250 (средняя длина фрагмента ДНК в ChIP-seq) и поделить на длину генома в bp. Если исследуется гистоновая модификация, то нужно оценить, сколько процентов генома занято ею. Эту информацию можно подчерпнуть из опубликованных экспериментов ChIP-seq, либо из результатов иммуноокрашивания хромосом.
Например, если в геноме дрозофилы 10000 сайтов связывания ТФ, то высадить получится не более 1.8% (10000*250/1.4x107) от стартового количества ДНК. Если на старте иммунопреципитации взято 10 мкг геномки, то получится осадить 180 нг ДНК. В реальности получиться может как меньше, так и больше. Во-первых, в ChIP-seq даже у хороших антител 30-70% всех фрагментов, выравненных на геном, оказываются за пределами сайтов связывания. Во-вторых, только некоторые локусы в геноме заняты белком во всех клетках. В-третьих, для полного осаждения ДНК необходимо подобрать правильное количество антител. Емкость антител может различаться между фасовками, и ее можно определить в контрольном эксперименте с заведомо превышенным количеством хроматина на старте.
В эксперименте с антителами против H3K27me3 от Abclonal (A22006, брали 1 мкг антител) нам удалось получить 300 нг ДНК в AB+ при стартовом количестве эмбрионов 25 и 50 мкл. Такого количества было достаточно для пробоподготовки, qPCR и даже для электрофореза.
- Какой выход ожидается в AB-?
В идеальном эксперименте в образце AB- должно выделяться минимальное количество ДНК (в 1000-100000 раз меньше, чем в AB+), либо вообще ничего. Это говорит о том, что магнитные частицы и пластик пробирки не взаимодействуют с хроматином. Если же в ходе эксперимента хроматин осаждается на шарах, то перед иммунопреципитацией необходимо провести дополнительную очистку хроматина шариками без антител. Для этого отмытые шарики (без антител) добавляют к фрагментированному хроматину на 1-3 часа, после чего шарики удаляются и ставится иммунопреципитация.
После преблокирования 10 мкл магнитных частиц Dinabeads Protein A+G c Blocking Buffer выход в AB- в эксперименте с 25-50 мкл эмбрионов был нулевым (количество ДНК было недостаточно для измерения на Qubit). Эксперименты без преблокирования шариков мы делали на мышиных ЭСК, но здесь выход в AB- был 1-2 нг на 500 тысяч клеток (~5 мкг хроматина).
- Какой выход ожидается в Input?
Чтобы понять это, перед началом эксперимента необходимо оценить стартовое количество ДНК. Например, путем предварительного эксперимента было посчитано, что из 15 мкл 12-часовых эмбрионов можно выделить 12 мкг геномной ДНК. Значит, если на эксперимент взяли 50 мкл эмбрионов, то на старте будет 40 мкг ДНК. Допустим, во время отмывок и фиксаций потери объема составили 10%. Значит, после фрагментации должно остаться около 36 мкг ДНК. Если весь хроматин разделили на AB+ и AB- поровну, а на Input взяли 10% от объема AB+, то в Input стоит ожидать ~1.5 мкг ДНК. Обычно, поскольку эффективность экстракции ядер во время отмывок ниже 100%, в ходе фрагментирования часть хроматина остается в осадке, а также из-за потерь во время преципитации нуклеиновых кислот выход ДНК в Input несколько ниже, так что высадить 50% от этого количества будет совершенно нормальным. Если же выход ниже 10%, то причиной может быть недофиксация, загрязнение нуклеазами и большие потери на каком-то из этапов работы. Все это может говорить о неудачном эксперименте ChIP.
В ходе иммунопреципитации H3K27me3 с ~50 мкл эмбрионов на старте мы выделили 1 мкг ДНК в Input. При количестве эмбрионов ~25 мкл было получено 500 нг.
- Сколько эмбрионов мне нужно взять на успешный эксперимент?
Данный протокол был опробован для эксперимента с иммунопреципитацией модифицированного гистона H3K27me3. Вполне достаточным оказалось взять 25 мкл эмбрионов. Правило, по которому выбирают стартовое количество материала, следующее: при изучении транскрипционных факторов, на один эксперимент (одна реплика и контроль AB-) берут эквивалент ~70 мкг ДНК (эквивалент 7 млн клеток человека). Для гистонов или белков, обладающих широким распространением по геному, количество стартового материала в 7-10 раз ниже (эквивалент 1 млн клеток человека).
- Я выделил ДНК и померил ее количество. Достаточно ли ее для пробоподготовки библиотек?
По-хорошему, для пробоподготовки требуется не менее 10 нг ДНК. В идеале, чем больше - тем лучше. Если выход в Input не является проблемой, то с AB+ все гораздо сложнее. Пользуясь данным протоколом, нам удавалось получать 300 нг на одну иммунопреципитацию с антителами против H3K27me3 (Abclonal). Однако нужно понимать, что большой выход может быть результатом сильного неспецифичного связывания антител. Исключить такую возможность можно с помощью qPCR с несколькими отрицательными контролями.
- У меня маленький выход ДНК в AB+, что мне делать?
Выход ДНК зависит главным образом от четырех факторов: содержания белка в хроматине, эффективности фиксации, количества стартового материала на момент добавления антител, и количества самих антител.
Если маленький выход не связан с низким количеством стартового материла, то причина может заключаться в недофиксации. Как правило, продолжительность и жесткость фиксации определяют в контрольном эксперименте. Важно понимать, что эксперименты с транскрипционными факторами, которые функционируют в составе мультисубъединичных комплексов требуют тщательной оптимизации на стадии кросслинкирования, поскольку недостаточная фиксация приведет к потере белка из комплексов с ДНК.
Количество антител для иммунопреципитации определяется их емкостью. Хорошие антитела обладают отличной емкостью (сотни нанограмм ДНК на мкг антител). В большинстве случаев выход сильно ниже. Тогда нужно добавить больше антител. Однако, если суммарная емкость антител превышает количество метки в образце, то это приводит к увеличению неспецифичных сигналов в qPCR и, в конечном итоге, в итоговом профиле ChIP-seq. Желательно провести предварительный эксперимент для определения правильного соотношения антител к хроматину.
- У меня нет праймеров для qPCR, где мне их найти?
Вариант номер один. Найти в интернете публикации, где делался ChIP-qPCR с выбранными антителами.
Вариант номер два. Найти в интернете опубликованные профили ChIP-seq. Можно обратиться, к базам данных GEO, ENCODE, modENCODE или NGSQC. По этим профилям определить локусы с самым высоким обогащением изучаемой метки, и локусы с самым низким. Затем подобрать праймеры на несколько локусов вручную с помощью Primer-BLAST
Вариант номер три. Обратиться к коммерчески доступным вариантам праймеров. Например, ActiveMotif предоставляет наборы праймеров для контроля результатов ChIP-seq. Последовательности праймеров заданы в сопроводительных документах.
Вариант номер четыре. В интернете можно найти базы данных, собирающие последовательности праймеров для ChIP или других экспериментов.
Нужно помнить, что некоторые факторы активно взаимодействуют с мобильными элементами. Праймеры на них могут быть хорошим контролем.
- Я выделил ДНК, но фрагменты длиннее 1 кб. Мой эксперимент испорчен?
Недостаточная фрагментация может возникнуть по нескольким причином. Во-первых, если концентрация хроматина на момент соникирования превышена, то эффективность фрагментации падает, и нужно больше циклов соникирования, чтобы добиться оптимального распределения фрагментов по длине. Во-вторых, сниженная эффективность фрамгентации может быть вызвана оверфиксацией. Возможно, стоит снизить продолжителность фиксации. Во-третьих, фрагментация эухроматина и гетерохроматина происходит с разной эффективностью, поскольку открытая ДНК гораздо легче разрывается ультразвуком. По этой причине иммунопреципитация белков, ассоциированных с гетерохроматином, приводит к осаждению фрагментов большей длины.
Фрагменты ДНК длинее 1 кб с большой вероятностью будут утрачены в ходе пробоподготовки, поэтому в ситуации, когда большая доля фрагментов лежит выше этой отметки, стоит перед пробоподготовкой провести дополнительный раунд соникирования. Подобрать нужную продолжительность соникирования можно в контрольном эксперименте с обработкой нескольких аликвот Input ультразвуком разной продолжительности.
- Я выделил ДНК, но фрагменты короче 100 пар. Мой эксперимент испорчен?
Слишком короткие фрагменты (<50 пн) будут потеряны при пробоподготовке. Кроме того, секвенировать короткие фрагменты может быть крайне расточительным, поскольку в настоящее время длина прочтений на разных секвенирующих платформах превышает 70 bp. Если длина прочтений сильно превышает длину фрагментов с адаптерами, то во-первых, полученные прочтения будут содержать последовательности адаптеров и их придется обрезать, чтобы нормально выровнить на геном (это расточительно), а во-вторых, если при запуске секвенатора много кластеров будут обрываться до завернения программы, это приведет к сбою работы секвенатора и испорченному запуску. Поэтому следует избегать секвенировать библиотеки со слишком коротким фрагментным составом.
Такой проблема может возникнуть по нескольким причинам: недофиксация, слишком большая продолжительность соникирования и загрязнение нуклеазами.
Причиной недофиксации может являться испорченный формальдегид (нужно убедиться, что в использованном формальдегиде не выпал осадок) либо слишком большое количество стартового материала.
Чтобы решить проблему оверфрагментации, нужно снизить продолжительность соникирования.
Загрязнение нуклеазами решается стандартными методами.
- Я сделал qPCR, все ли у меня в порядке?
В проверочном qPCR определяется выход ДНК в AB+ и AB- относительно максимально возможного. Максимальный выход измеряется при помощи Input. Если объем Input составлял 10% от AB+ (и AB-), то концентрацию ДНК в Input по каждому локусу делят на 10/100. Это и будет максимальный выход, который вы ожидали.
В эксперименте используют праймеры, соответсвующие локусам с высоким обогащением исследуемого фактора (известный сайт связывания ТФ, или локус внутри домена H3K27me3). В качестве негативного контроля используются праймеры на локусы, в котором метка отсутствует (антипики в экспериментах ChIP-seq).
В AB+ выход в “+”-локусах не обязательно должен быть максимальным. Строго говоря, если количество хроматина в эксперименте превышало емкость антител, то получить максимальный выход невозможно. По этой причине нужно в первую очередь ориентироваться на соотношение сигнала в “+” и “-”-локусах. Чем оно выше - тем более высокие отличия между специфичным и неспецифичным сигналом будут получен в эксперименте. Строго говоря, это отношение является показателем качества антител. Для хороших антител это соотношение должно превышать 5-10 раз. На профилях ChIP-seq это отношение можно увидеть, сравнив высоту пиков в “+” и “-”-локусах.
Увеличенный сигнал в негативном контроле может говорить об избытке антител.
Важно понимать, что распределение метки в геноме сильно зависит от типа клеток, генотипа и условий содержания. По этой причине не стоит отчаиваться, если негативный контроль дает повышенный сигнал. Перед тем как отбраковывать эксперимент, нужно проверить, может ли повышенный сиганл в “-”-локусе быть следствием биологических процессов. Чтобы понять это, стоит подбирать для проверки не один “-”-контоль, а несколько.
В образце AB- сингал в “+” и “-” локусах предупреждает о наличии фона на будущем профиле ChIP-seq (низкое отношение сигнал/шум). Высокий фон приводит к низкой разрешающей способности ChIP-seq, к потере сайтов связывания или появлению ложно-положительных сайтов. Фактически, на “шумных” профилях ChIP-seq сайты связывания с низким уровнем обогащения неразличимы на окружающем фоне, а локусы с большим уровнем шума, наоборот, будут выглядеть как сайты связывания. Как правило, уровень фона на зависит от положения в геноме, так что в AB- в “+” и “-”-локусах виден приблизительно одинаковый уровень обогащения. Если уровень обогащения в AB- равен 10%, то в ChIP-seq профиле сайты связывания с аналогичным уровнем обогащения будут неразличимы на общем фоне. Более того, сайты, в которых исследуемая метка отсутсвует, будут демонстрировать такой же уровень обогащения. В хороших экспериментах, уровень фона должен быть на 3-4 порядка ниже уровня сигнала в “+”-контроле AB+ или, еще лучше, вообще отсутсвовать.
Высокий фон возникает из-за интенсивных неспецифических взаимодействий между компонентами хроматина и поверхностью магнитных частиц или пластика. Уменьшить фон можно различными способами: понизив стартовое количество или концентрацию хроматина при иммунопреципитации, уменьшив количество шариков, блокированием шариков с BSA, преинкубацией хроматина на магнитных шарах без антител, увеличив количество отмывок, увеличив концентрацию солей в отмывочных буферах. Судя по опыту, магнитные частицы Dinabeads Protein A+G плохо взаимодействуют с хроматином эмбрионов, особенно после преблокирования. Поэтому сильных проблем здесь не должно возникнуть.
- Я хочу отсеквенировать образцы и уже выделил ДНК, что делать дальше?
Если выход иммунопреципитации составляет ~100 нг и больше, то для пробоподготовки можно использовать наборы по типу TruSeq Nano DNA. Если выход составляет от 1 до 10 нг, желательно пользоваться наборами Nextera.
Если сделать пробоподготовку библиотек своими силами не вариант, то можно обратиться к коммерческим организаций. Например, услуги по пробоподготовке предлагает Геномед.
Источники
-
Протокол Loubiere et al. 2017 ChIP-seq цельных эмбрионов дрозофилы. Обратите внимание, что в протоколе немного отличаются составы буферов, кроме того, здесь дается подробное описание процедуры сбора эмбрионов.
-
Протокол Loser et al. 2016.
-
Протокол Cavalli et al. 1999.
-
Развернутые инструкции по оптимизации ChIP от Cell Signals.
-
Иммунопреципитация хроматина: оптимизация, количественный анализ и нормализация данных Haring et al. 2007.
-
Рекоммендации abcam, Merk и Creative Biolabs по исправлению проблем.