Bowtie2
Bowtie 2 — это наиболее популярный инструмент для выравнивания коротких фрагментов ДНК на эталонные последовательности. Он особенно хорош для выравнивания ридов длиной от 50 до 100-1000bp на длинные референсные геномы (в т.ч. млекопитающих). Достаточно высокая скорость и эффективное использование оперативной памяти в Bowtie 2 достигается за счет преобразования генома в матрицу ссылочных последовательностей с помощью FM-индекса (на основе преобразования Барроуза — Уилера, Burrows-Wheeler transform. Bowtie2 поддерживает режимы выравнивания с пропусками (gapped alignment), локальное выравнивание (local alignment) и выравнивание парных ридов (paired-end alignment). Доступен для Linux и OSX.
Не следует путать bowtie2 с bowtie. Хотя последний инструмент является предшественником bowtie2, в нем имеется ряд недостатков: отсутствует возможность локального выравнивания (то есть все риды должны выравниваниваться полностью от начала до конца), режим выравнивания парных ридов ограничен и не предназначен для выравнивания дискордантных ридов, отсутсвует поддержка вырожденных символов (например, N). Bowtie обладает низкой эффективностью при выравнивании ридов длинее 50 пар и не способен выравнивать риды длинее 1000 пар. Важно понимать, что геномные индексы, произведенные Bowtie2 и Bowtie взаимозаменяемы только начиная с версии Bowtie v1.2.3.
Референсные публикации
-
Langmead B, Wilks C, Antonescu V, Charles R. Scaling read aligners to hundreds of threads on general-purpose processors. Bioinformatics. 2018 Jul 18. doi: 10.1093/bioinformatics/bty648.
-
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature Methods. 2012 Mar 4;9(4):357-9. doi: 10.1038/nmeth.1923.
Официальные материалы
-
Расширенный мануал
-
Список приложений, использующих Bowtie2
-
Архив геномных индексов Bowtie2 и Bowtie
Принцип работы
Подробно о алгоритме преобразования BWT, FM-индексе и связанных с этим алгоритмах можно познакомиться в видео-лекции, презентации или конспекте.
Если говорить подробно, то для каждого рида Bowtie2 производит вычисления в четыре шага. На шаге 1 (Extract seed) Bowtie2 извлекает из рида и комплементарной к нему последовательности (не путать с mait-pair) “затравочные” подстроки (seeds). На шаге 2 (Align with FM-Index) затравочные подстроки выравниваются на референсный геном без пропусков (ungapped, без делеций и инсерций) с использованием FM-индекса, в результате чего получаются интервалы Барроуза-Уиллера (BW ranges), которые указывают на строки в матрице, полученной путем трансформации генома с помощью BWT. На шаге 3 (Prioritize, resolve) Bowtie2 извлекает из BW-матрицы строки, соответствующие полученным интервалам, и каждая строка приобретает приоритет (prioritize), при этом чем меньше интервал - тем выше приоритет. Далее, учитывая приоритет строк, с помощью FM-индекса и алгоритма “walk-left” Bowtie2 определяется смещение (offset) выбранных строк, и таким образом получает координаты затравочных последовательностей в геноме (Extention candidates). Иными словами, Bowtie2 разрешает (resolve) затравочные выравнивания. На шаге 4 (Extend) Bowtie2 берет приоритетные разрешенные затравочные выравнивания из шага 3 и расширяет выравнивание рида на всю окрестность вблизи затравки. Выравнивание на данном этапе ускоряется за счет использования динамического программирования SIMD (Single Instruction Multiple Data). Выравнивание продолжается до тех пор, пока не будут проанализированы все разрешенные затравки (seed hits), пока не будет изучено достаточное количество выравниваний, или пока не будет достигнут предел трудоемкости (effort limit) динамического программирования.
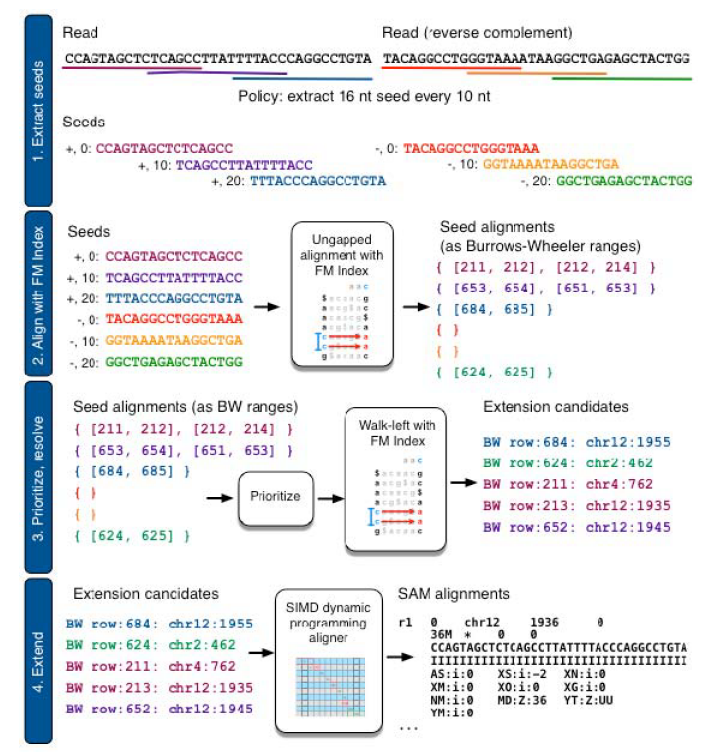
Если кратко, то сначала каждый рид и комплементарная к нему последовательность разбивается на затравочные подстроки (seed). Затем извлеченные подстроки выравниваются на референсный геном без пропусков (seed alignment) и расставляются по приоритету (prioritize). На последнем этапе затравочные выравнивания расширяются на весь рид.
В результате для каждого рида получается множество выравниваний. Чтобы выбрать наилучшее выравнивание Bowtie2 вычисляет для каждого из них оценку выравнивания (alignment score). Оценка количественно определяет, насколько рид похож на референсную последовательность. Чем выше балл - тем лучше. Оценка рассчитывается путем вычитания штрафов (penalty) за каждое отличие: замены (mismatch), пропуски (gap), наличие вырожденных символов (ambigous characters). В режиме локального выравнивания, за каждое совпадение к оценке добавляется бонус (bonus). Результат выравнивания оформляется в формате SAM в поток стандартного вывода 1 (“stdout”), если не указан файл назначения. Дополнительная информация, предупреждения и статистика выравнивания выводятся в стандартный поток номер 2 (“stderr”).
Установка
Подробное описание способов установки можно посмотреть здесь
Наиболее простой способ - установить с помощью Anaconda:
conda install -c bioconda bowtie2
Инструменты Bowtie2
После установки Bowtie2 пользователю становится доступно несколько инструментов:
bowtie2,bowtie2-align-sиbowtie2-align-lосуществляют выравнивание коротких последовательностей (в формате fastq, fasta и др.) на индексированный референсный геном/последовательность.bowtie2-align-sиbowtie2-align-lпредставляют собой бинарный код для выравнивания на индексы малого (small) и большого (large) формата.bowtie2является сценарием-оберткой (wrapper), запускающимbowtie2-align-s/lв зависимости от формата индекса и дающий дополнительные возможности, в частности, работу со сжатыми файлами. По-сути всегда нужно использоватьbowtie2.bowtie2-build,bowtie2-build-s, иbowtie2-build-lосуществляют индексирование генома (файл в формате fasta или последовательности из командной строки). Как и в прошлом пункте,bowtie2-buildпредставляет собой обортку для запуска бинарных командbowtie2-build-s, иbowtie2-build-l. Для геномов длиной менее 4 миллиардов нуклеотидов bowtie2-build строит “малый” индекс, используя 32-битные числа в различных частях индекса. Если геном длиннее,bowtie2-buildстроит “большой” индекс, используя 64-битные числа. Малые индексы хранятся в файлах с расширением .bt2, а большие - в файлах с расширением .bt2l. Пользователю не нужно беспокоиться о том, является ли конкретный индекс малым или большим, так как обертки автоматически построят и используют соответствующий индекс.bowtie2-inspect,bowtie2-inspect-sbowtie2-inspect-lизвлекают из индекса Bowtie2 информацию о том, что это за индекс и какие референсные последовательности использовались для его построения. При запуске без каких-либо параметров инструмент выведетFASTAфайл, содержащий последовательности референсов (все символы кромеA/C/G/Tбудут преобразованы вN).
Индексирование генома с помощью bowtie2-build
Готовые геномные индексы можно скачать с оффициального сайта bowtie-bio.sourceforge.net
Обычно в качестве референса предоставляется fasta-файл. Вслед за fasta-файлом требуется префикс для файлов индексированного референса:
bowtie2-build -f путь_к_fasta Префикс
Параметр -f не обязателен. Он указывает, что рефернс представляет из себя fasta-файл. Можно предоставлять список fasta-файлов через запятую без пробелов:
bowtie2-build -f путь_к_fasta_1,путь_к_fasta_2,путь_к_fasta_3 Префикс
Вместо Fasta-файла может быть список последовательностей через запятую без пробелов:
bowtie2-build -c GGTCATCCT,ACGGGTCGT,CCGTTCTATGCGGCTTA Префикс
В результате программа выдаст шесть файлов с названиями Префикс.1.bt2, Префикс.2.bt2, Префикс.3.bt2, Префикс.4.bt2, Префикс.rev.1.bt2 и Префикс.rev.2.bt2. Если префикс не указать, то префикс будет NAME.. В случае, если индексы большие, вместо .bt2 расширение файла будет .bt2l.
Файлы индексированного референса будут сохранены в директории, из которой была вызвана программа bowtie2-build
Для ускоренного индексирования следует активировать многопоточный режим, добавив параметр --treads n.
bowtie2-build --threads 25 -f путь_к_fasta Префикс
Множественное выравнивание с помощью bowtie2
Минимальный вид команды:
bowtie2 -x <bt2-idx> {-1 <m1> -2 <m2> | -U <r> } -S [<sam>]
Для выравнивания нужно обязательно указать путь к индексированному референсу после параметра -x. Это путь к папке, в которой содержатся файлы индекса плюс префикс файлов-индексов. Например:
-x /home/romanov/DM6_drosophila/DM6
Для выравнивания односторонних прочтений (single-end reads) нужно указать полный путь к файлу fastq (или fastq.gz) после параметра -U:
-U /home/file.fastq.gz
Также можно указать несколько fastq-файлов через запятую:
-U /home/file1.fastq.gz,../file2.fastq.gz,~/foldeir2/file3.fastq.gz
Для выравнивания двусторонних прочтений (paired reads) нужно указать полный путь к файлам R1 и R2 после параметров -1 и -2:
-1 /home/file.R1.fastq.gz -2 /home/file.R2.fastq.gz
Также можно указать несколько fastq-файлов через запятую:
-1 /home/file1.R1.fastq.gz,../file2.R1.fastq.gz -2 /home/file1.R2.fastq.gz,../file2.R2.fastq.gz
По умолчанию результат выравнивания в формате .SAM выводится в стантартный вывод (экран командной строки). Чтобы вывести результат в файл, нужно указать адрес после параметра -S:
-S путь_к_файлу/alignment.sam
Режимы выравнивания
Bowtie2 способен выравнивать последовательности на индекс в двух режимах:
- В режиме end-to-end (сквозное выравнивание) bowtie2 ищет в референсе участок, идентичный целевой последовательности от начала до конца. Такое выравнивание также называют необрезанным (“unclipped” или “untrimmed”). Bowtie2 осуществляет сквозное выравнивание по умолчанию.
- В режиме local (локальное выравнивание) bowtie2 ищет в референсе участок, идентичный райну внутри целевой последовательности, при этом несколько нуклеотидов на концах последовательности не учитываются, то есть мягко обрезаются (“soft-clipping” или “soft-trimming”). Это значит, что несовпадение концевых нуклеотидов в последовательности не учитывается
Пример сквозного выравнивания:
Рид: GACTGGGCGATCTCGACTTCG
||||| |||||||||| |||
Референс: GACTG--CGATCTCGACATCG
Пример локального выравнивания:
Рид: ACGGTTGCGTTAA-TCCGCCACG
||||||||| ||||||
Референс: TAACTTGCGTTAAATCCGCCTGG
Виды несовпадений
Замены (mismatches)
Рид: GACTGCGCCGATTTCGACTTCG
|||||| ||||| |||||||||
Референс: GACTGCCCCGATCTCGACTTCG
Вырожденные буквы (ambigous characters)
Рид: GACTGCGCCGATNTCGACTTCG
|||||| ||||| |||||||||
Референс: GACTGCNCCGATCTCGACTTCG
Пропуск внутри референса (reference gap)
Рид: GACTGGGGCGATCTCGACTTCG
||||| ||||||||||||||
Референс: GACTG---CGATCTCGACTTCG
Пропуск внутри рида (read gap)
Рид: GACTG---CGATCTCGACTTCG
||||| ||||||||||||||
Референс: GACTGGGGCGATCTCGACTTCG
Функции
Большинство параметров bowtie2 принимают на входе либо число, либо строку. Однако есть параметры, которые принимают функции (functions) в форматеA*F(X)+B. Функции записываются следующим образом: –параметр <F>,<B>,<A>.
Символ F обозначает название функции. Всего функции бывают четырех типов: константа (C), линейная функция (L), квадратный корень (S), натуральный логарифм (G).
Например, параметр записанный в виде C,2,2 означает константу F(X)=2+2=4.
Запись L,-5,-10.1 означает линейную функцию F(X)=-10.1*X-5.
Функция, записанная в виде S,-0.6,8 означает функцию F(X)=8*sqrt(X)-0.6.
Функция, записанная в виде G,3,-6 означает функцию F(X)=-6*ln(x)+3.
Оценка выравнивания
Каждая последовательность может выравниваться на референс в нескольких местах. Bowtie2 определяет для каждого выравнивания величину оценки (alignment score) и на выходе выдает координаты выравнивания с оценкой, превышающей пороговое значение (minimum score threshold). При сквозном выравнивании оценка выравнивания вычисляется путем вычитания штрафов (penalties) за каждое отличие в последовательности или референсе (замены, пропуски и т. д.). При локальном выравнивании за каждый совпавший нуклеотид к оценке добавляется бонус (bonus).
Ниже приведены параметры, которые задают значения бонусов, штрафов и пороговой оценки:
--ma <целое число X> - бонус за совпадение (match bonus). Используется только при локальном выравнивании (иначе выдаст ошибку). По-умолчанию равно 2. Если рид состоит из 50 нуклеотидов и все нулкеотиды совпадают с референсом, то оценка будет равна X*50=100
--mp <целое число MN>,<целое число MX> - штраф за замену (mismatch penalty). Требует два числа, введенных через запятую. Число MN - это минимально-возможный штраф, MX - максимально возможный. Величина штрафа зависит от качетсва прочтения в несовпадающем нуклеотиде (Phred-score). Если Phred-score равен Q, то от оценки отнимается число равное целой части от величины MN+(MX-MN)*(min(40,Q)/40). Так что если Q>40, то штраф будет равен MX, если Q равен 0, то штраф будет равен MN. По умолчанию: MX=6, MN=2. Можно ввести параметр --ignore-quals, тогда всегда будет отниматься MX.
--np <целое число> - Устанавливает штраф за позиции в выравнии, где рид или референс, или оба сразу содержат неоднозначный символ, например N. По умолчанию: 1.
--rdg <целое число N1>,<целое число N2> - Штраф за пропуски (read gaps) внутри рида. Если пропуск перекрывает только один нуклеотид, то штраф равен N1. Штраф за пропуск M нуклеотидов подряд составляет N1 + M*N2. Таким образом, N1 - это штраф за открытие пропуска (gap open), а N2 -это штраф за расширение пропуска (gap extend). По умолчанию: N1=5, N2=3.
--rfg <целое число N1>,<целое число N2> - Штраф за пропуски внутри референса (reference gaps). Штраф за пропуск M нуклеотидов подряд составляет N1 + M*N2. По умолчанию: N1=5, N2=3.
--score-min <функция> - Задает функцию, управляющую минимальной оценкой выравнивания, необходимой для того, чтобы выравнивание считалось «действительным» (valid), то есть достаточно хорошим. Это функция от длины рида. Значение по умолчанию в сквозном режиме L,-0.6,-0.6, а в локальном режиме G,20,8.
Число выравниваний для каждого рида
По умолчанию bowtie2 для каждого рида находит одно действительное выравнивание, после чего ищет еще выравнивания с такой же или более высокой оценкой. В конце концов bowtie2 докладывает одно выравнивание с наибольшей оценкой, или если выравниваний с идентичной оценкой несколько, выдает одно случайное. Для некоторых задач требуется узнать все возможные выравнивания для рида (например, картирование повторов в геноме). Для этого нужно ввести параметры -k или -a. Оба параметра замедляют процесс выравнивания.
-k <целое число> - это число выравниваний, которое будет записано в SAM-файл для каждого рида.
-a - эта команда заставляет bowtie2 записать все выравнивания с оценкой качества, превышающей порог.
Cлучайный режим
В тех случаях, когда bowtie2 приходится выбирать для рида одно выравнивание из нескольких вариантов с одинаковой оценкой качества, он использует генератор случайных чисел. В результате, разные запуски программы на одних и тех же данных приводят к неэксвивалентным результатам. Чтобы получать одинаковые результаты, нужно при запуске программе передать на вход программе одно и то же начальное значение генератора случайных чисел при помощи параметра --seed <целое число>.
Если же в fastq-файлах есть много повторяющихся ридов и требуется, чтобы каждый рид выравнивался в случайном месте генома, то нужно использовать параметр --non-deterministic, который заставляет программу запускать генератор случайных чисел для каждого рида отдельно. При этом начальным значением является время на часах.
Выравнивание парных ридов
При выравнивании спаренных ридов (mate pairs) положение ридов относительно референса зависит от метода пробоподготовки. В SAM-файле Bowtie2 помечает спаренные риды разными способами, указывая, были риды выравнены правильным образом. Чтобы определить, правильное выравнивание, bowtie2 учитывает длину встройки (максимальна дистанция между концами ридов), ориентацию ридов относительно прямой цепи референса, положение первого и второго ридов относительно друг друга. Конкордантными парами называются пары ридов, которые выравниваются на одну и ту же хромосому (или скэффолд), имеют правильную ориентацию и взаимное положение, а также правильную дистанцию. Спаренные риды, которые не удовлетворяют условиям называют дискордантными.
Минимальная длина фрагментов указывается параметром -I <целое число> или --minins <целое число> (это короткое и длинное название параметра). По умолчанию это значение равно 0, то есть минимум не установлен.
Максимальная длина фрагментов указывается параметром -X <целое число> или --maxins <целое число> . По умолчанию значение равно 500 (пятьсот пар нуклеотидов). Чем больше разница между -X и -I, тем медленнее работает программа!
Параметры --fr/--ff/--rf указывают возможную ориентацию первого и второго рида относительно прямой цепи референса (см. картинку ниже). Если ориентация ридов не удовлетворяет выбранному условию, то рид считается дискордантным.
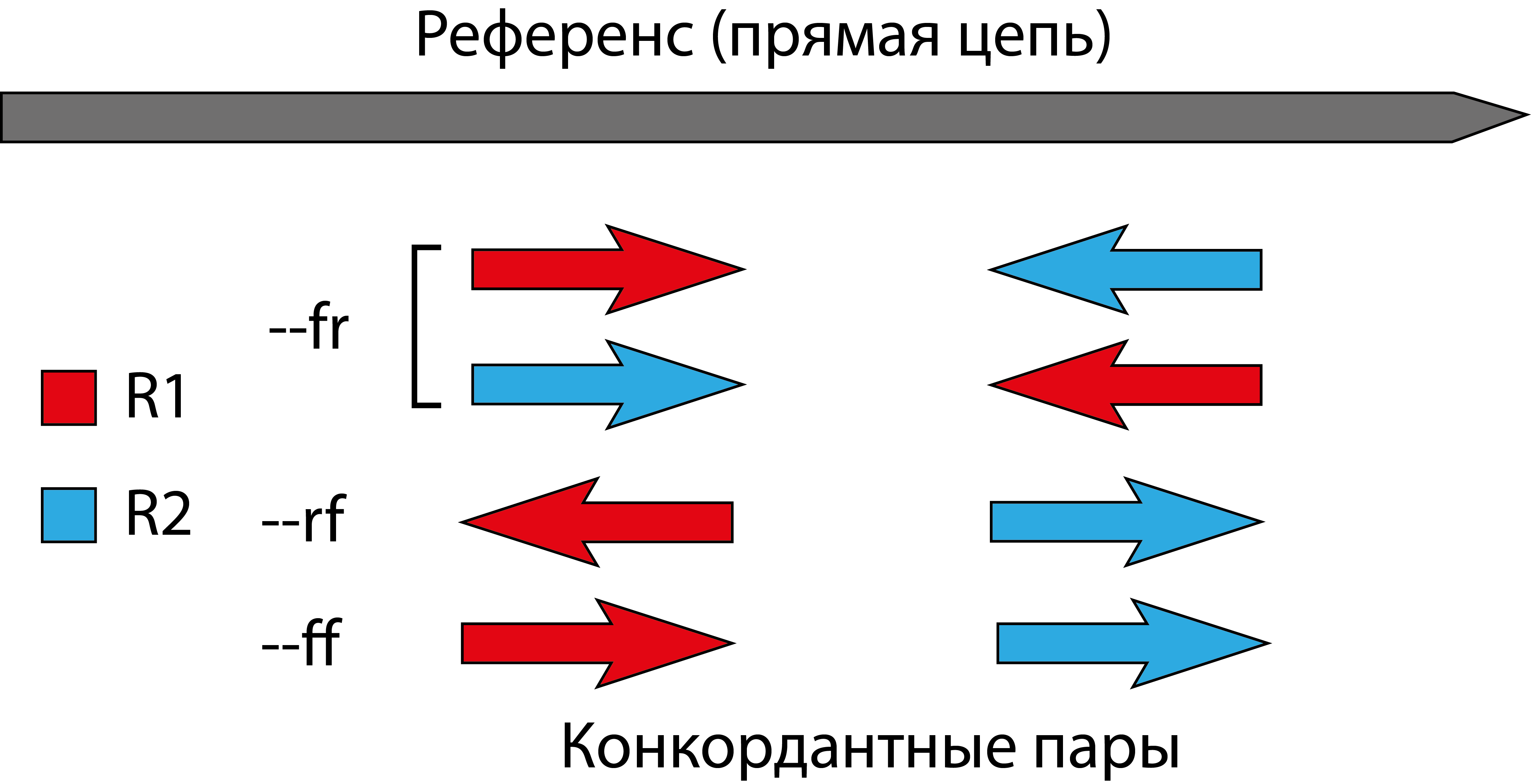
Если риды перекрываются, то bowtie2 учитывает тип перекрывания, когда классифицирует конкордантные и дискордантные пары.
Спаренные риды могут перекрываться обычным способом (overlap). Такие пары считаются конкордантными. Но использование параметра --no-overlap превращает такие риды в дискордантные:
Пара 1: GCAGATTATATGAGTCAGCTACGATATTGTT
Пара 2: TGTTTGGGGTGACACATTACGCGTCTTTGAC
Референс: GCAGATTATATGAGTCAGCTACGATATTGTTTGGGGTGACACATTACGCGTCTTTGAC
Содержать друг друга (contain). Такие пары считаются конкордантными, а параметр параметр --no-contain заставляет считать такие пары дискордантными:
Пара 1: GCAGATTATATGAGTCAGCTACGATATTGTTTGGGGTGACACATTACGC
Пара 2: TGTTTGGGGTGACACATTACGC
Референс: GCAGATTATATGAGTCAGCTACGATATTGTTTGGGGTGACACATTACGCGTCTTTGAC
Пара 1: CAGCTACGATATTGTTTGGGGTGACACATTACGC
Пара 2: CTACGATATTGTTTGGGGTGAC
Референс: GCAGATTATATGAGTCAGCTACGATATTGTTTGGGGTGACACATTACGCGTCTTTGAC
Или “проходить мимо” друг друга. Такое перекрывание называется «ласточкин хвост» (dovetail) и по умолчанию считается дискордантным:
Пара 1: GTCAGCTACGATATTGTTTGGGGTGACACATTACGC
Пара 2: TATGAGTCAGCTACGATATTGTTTGGGGTGACACAT
Референс: GCAGATTATATGAGTCAGCTACGATATTGTTTGGGGTGACACATTACGCGTCTTTGAC
Перекрывание по типу ласточкин хвост можно заставить считать конкордантным, если ввести параметр --dovetail.
По умолчанию bowtie2 для начала ищет конкордантные выравнивания, и только если таких не находится, приступает к поиску дискордантных. Чтобы запретить вывод дискордантных ридов, нужно ввести параметр --no-discordant.
Когда bowtie2 не может найти конкордантные и дискордантные выравнивания для спаренных ридов, он ищет отдельные независимые выравнивания для каждого рида в отдельности (например, пытается выравнивать риды на разные хромосомы). Если такое поведение неприемлимо, то нужно ввести параметр --no-mixed.
Параметры выравнивания
Чтобы быстро сузить количество возможных выравниваний, которые необходимо учитывать, bowtie2 начинает с извлечения затравночных подстрок из прочтения и комплементарной к нему последовательности и выравнивания их без пропусков с помощью индекса FM. Этот начальный шаг (многозатравочная эвристика, multiseed euristic) делает Bowtie 2 намного быстрее, чем без такого фильтра, но за счет пропуска некоторых допустимых выравниваний. Например, рид может иметь правильное общее выравнивание, но не иметь допустимых зерновых выравниваний, потому что каждое потенциальное зерно прерывается слишком большим количеством замен или пробелов.
Компромисс между скоростью и чувствительностью/точностью можно отрегулировать, установив длину начальной затравки (-L), дистанция между затравками (-i)допустимое количество замен на затравку (-N). Для более точного выравнивания можно: (а) уменьшить дистанцию между затравками, (б) сократить длину затравок и/или (в) разрешенить большее количество замен.
-N <целое число> определяет число замен в затравочных выравниваниях. По умолчанию - 0. Может принимать значение 0 и 1.
-L <целое число> определяет длину затравночной полседовательности. В end-to-end режиме равно 22, а в local 20.
-i <целое число> - функция, связывающая длину прочтения и расстояние между затравочными последовательностями. По умолчанию равно S,1,1.15 в --end-to-end режиме и S,1,0.75 в --local mode.
Есть еще два параметра, контролирующих скорость/чувствительность выравнивания:
-D <целое число X> - перед тем как сдвинують затравочную последовательность вдоль референса, bowtie2 расширяет затравку с целью получить более качественное выравнивание. Если после расширения затравки качество выравнивания не растет, то bowtie2 увиличивает счетчик на единицу. А число X определяет максимальное число, которое может достигнуть счетчик для затравочной последовательности. Увеличение -D делает Bowtie 2 медленнее, но увеличивает вероятность того, что он сообщит правильное выравнивании для ридов, которые выравниваются на много мест в референсе. По умолчанию это число равно 15.
-R <целое число X> - максимальное число раз, когда bowtie2 перевыравнивает прочтение (re-seed), если прочтение имеет несколько выравниваний в геноме. Чем больше число, тем медленнее выравнивание, но тем больше вероятность найти правильное выравнивание. Значение по умолчанию - 2.
--n-ceil <функция> - устанавливает функцию, связывающую максимальное число вырожденных букв в выравнивании в зависимости от длины рида. По умолчанию: L,0,0.15.
--local - включает локальный режим выравнивания, при котором несколько нуклеотидов на концах прочтения обрезаются и не вносят вклад в качество выравнивания.
--end-to-end - включает сквозной режим выравнивания. Этот параметр включен по умолчанию.
--gbar <целое число> - определяет число нуклеотидов на концах рида, между которыми недопустимы пробелы (gaps). По умолчанию 4.
--ignore-quals - заставляет bowtie2 игнорировать Phred-score при подсчете пенальти. В таком случае штраф за каждую замену максимальный.
--no-1mm-upfront - По умолчанию, чтобы увеличить скорость выравнивания bowtie2 для начала ищет для каждого рида выравнивание с полным совпадением или с одной ошибкой. Это гораздо быстрее, чем применить многозатравочную эвристику. В случае, если длина затравки (-L) выбрана равной длине рида, а число замен (-N) равно нулю, то такой подход приведет к появлению выравниванию с заменами. Данный параметр отключает такое поведение.
Чтобы не менять параметры каждый раз, авторы сделали пресеты параметров мультизатравочного выравнивания, которые различаются скоростью и чувствительностью: Вариант end-to-end:
--very-fast - То же, что и : -D 5 -R 1 -N 0 -L 22 -i S,0,2.50
--fast - То же, что и : -D 10 -R 2 -N 0 -L 22 -i S,0,2.50
--sensitive - То же, что и : D 15 -R 2 -N 0 -L 22 -i S,1,1.15 (по умолчанию в --end-to-end)
--very-sensitive - То же, что и: -D 20 -R 3 -N 0 -L 20 -i S,1,0.50
Вариант local:
--very-fast-local - То же, что и: -D 5 -R 1 -N 0 -L 25 -i S,1,2.00
--fast-local - То же, что и: -D 10 -R 2 -N 0 -L 22 -i S,1,1.75
--sensitive-local - То же, что и: -D 15 -R 2 -N 0 -L 20 -i S,1,0.75 (по умолчанию в --local)
--very-sensitive-local - То же, что и: -D 20 -R 3 -N 0 -L 20 -i S,1,0.50
Параметры ввода
-q - риды в формате fastq/fq
-f - риды в формате fasta/fa/mfa/fna
-c - риды записаны в командной строке как разделенный запятой список последовательностей
-s <целое число> или --skip <целое число> - пропустить первые несколько ридов в файле.
-u <целое число> или --qupto <целое число> - выровнить первые несколько ридов. Удобно для проверки кода.
-5 <целове число> или --trim5 <целое число> - перед выравнивание исключить из рида указанное число нуклеотидов с левого конца (включая спаренные риды). Полезно, так как позволяет избежать тримминга ридов.
-3 <целове число> или --trim3 <целое число> - перед выравниванием исключить из рида указанное число нуклеотидов с правого конца (включая спаренные риды). Полезно, так как позволяет избежать тримминга ридов.
--trim-to [5:|3:]<целое число> - перед выравниванием укоротить рид до указанной длины. Удаляться будут буквы с левого конца (--trim-to 5:N) или c правого конца (--trim-to 3:N). Нельзя вводить одновременно с параметром -5 или -3.
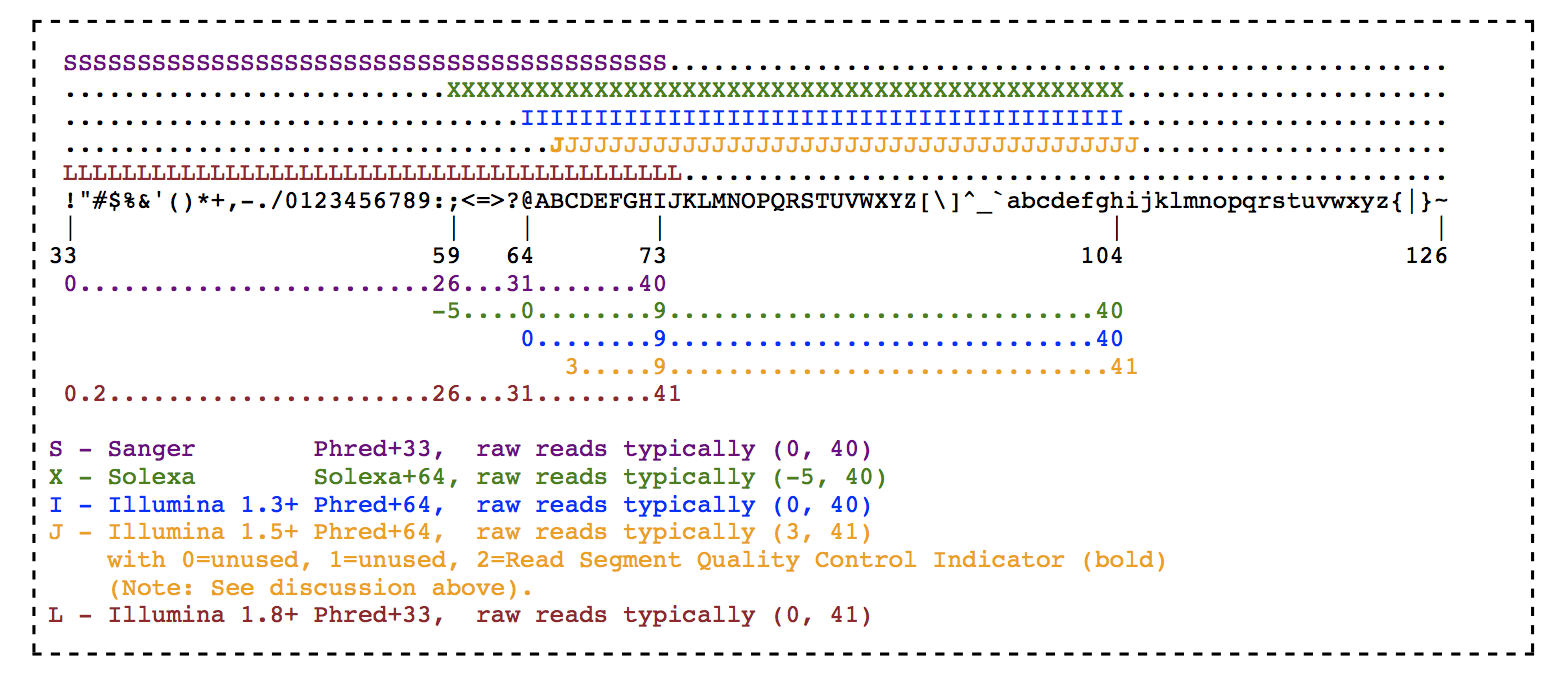
--phred33 или --phred64 - кодировка Phred-score в таблице ASCII. Зависит от платформы секвенирования. В Illumina MiSeq - phred33. Тип платформы можно опрделенить при помощи fastqc. Соответствие между платформой и Phred-score можно определить по картинке ниже:
Параметры вывода
|
Записать неспаренные невыравненные риды в файл <path> в зжатом или несжатом виде. |
|
Записать неспаренные риды, которые выравнились хотя бы один раз в файл <path> в сжатом или несжатом виде. |
|
Записать спаренные риды, которые не выровнились конкордантно, в файл <path> в зжатом или несжатом виде. |
|
Записать спаренные риды, которые выравнились конкордантно хотя бы один раз в файл <path> в сжатом или несжатом виде. |
Быстродействие
-p <целое число> или --threads <целое число> устаналвивает число занятых процессоров (по умолчанию 1). Ускоряет выравнивание в пропорциональное число раз.
Сводка по выравниванию
Сводка (Alignment summary) выглядит следующим образом:
20000 reads; of these:
20000 (100.00%) were unpaired; of these:
1247 (6.24%) aligned 0 times
18739 (93.69%) aligned exactly 1 time
14 (0.07%) aligned >1 times
93.77% overall alignment rate
А для спаренных ридов вот так:
10000 reads; of these:
10000 (100.00%) were paired; of these:
650 (6.50%) aligned concordantly 0 times
8823 (88.23%) aligned concordantly exactly 1 time
527 (5.27%) aligned concordantly >1 times
----
650 pairs aligned concordantly 0 times; of these:
34 (5.23%) aligned discordantly 1 time
----
616 pairs aligned 0 times concordantly or discordantly; of these:
1232 mates make up the pairs; of these:
660 (53.57%) aligned 0 times
571 (46.35%) aligned exactly 1 time
1 (0.08%) aligned >1 times
96.70% overall alignment rate
По умолчанию сводка выводится в стандартный поток ошибок stderr. Чтобы записать сводку в отдельный файл нужно воспользоваться перенаправлением из стандартного потока ошибок. Для этого в конце команды надо написать “2 > название_файла “:
bowtie2 -x /home/romanov/DM6_drosophila/DM6 -U file.fastq.gz --very-fast -S alignment.sam 2>alignment_summary.txt
Выравнивание сплайсированных прочтений RNA-seq
Bowtie2 используется некоторыми людьми для выравнивания ридов RNA-seq. С одной стороны, для этого в качества референса можно использовать fasta-файл с последовательностями транскриптов. С другой стороны, можно использовать bowtie2 в режиме --local и -k, чтобы найти все позиции внутри гена, куда выравнивается один рид. Преимуществом bowtie2 является скорость: судя по тестам, он может в 2 раза обыгрывать самые быстрые элайнеры RNA-seq, такие как Hisat2 или STAR. Важно понимать, что Bowtie2 предназначен для непрерываного выравнивания прочтений, так что при анализе резульлтатов такого выравнивания нужно четко отдавать себе отчет о возможных недостатках и трудностях.