STAR
Назначение STAR
Процесс выравнивания отсеквенированных последовательностей состоит из выбора подходящего эталонного генома для картирования полученных прочтений и выравнивания прочтений с использованием одного из нескольких инструментов. В отличие от других ChIP-seq или секвенирования генома, прочтения RNA-seq содержат сплайсированные последовательности. Это означает, что прямое сопоставление прочтений с референсом путем выравнивания ридов по всей длине сделает невозможным картирование большинства фрагментов. Это свойство транскриптов учитывается несколькими элайнерами, среди которых наиболее популярны STAR, HISAT2 или TopHat. Выбор элайнера часто является личным предпочтением, а также зависит от доступных вам вычислительных ресурсов.
STAR стал популярным по нескольким причинам. Во-первых, это один из самых быстрых элайнеров. Во-вторых, точность выравнивания с помощью STAR в ряде тестов превышает конкурентов (не всегда). Пожалуй, по производительности он может уступать только Hisat2 (и то не всегда). Однако в отличие от последнего он предоставляет подробный отчет о положении сайтов-сплайсинга, может сразу подсчитывать число прочтений в транскриптах, строит профиль покрытия генома прочтениями (signal file), делает анализ нуклеотидных вариантов в прочтениях, оптимизирован для анализа single cell RNA-seq и пр. STAR активно поддерживается разработчиками, благодаря чему в нем постоянно появляются новые функции. Его ключевым недостатком является высокая требовательность к оперативной памяти. В тестах его требования к памяти превышают таковые у Hisat2 в 5-10 раз (20-40 Gb оперативной памяти при анализе генома человека).
Описание алгоритма
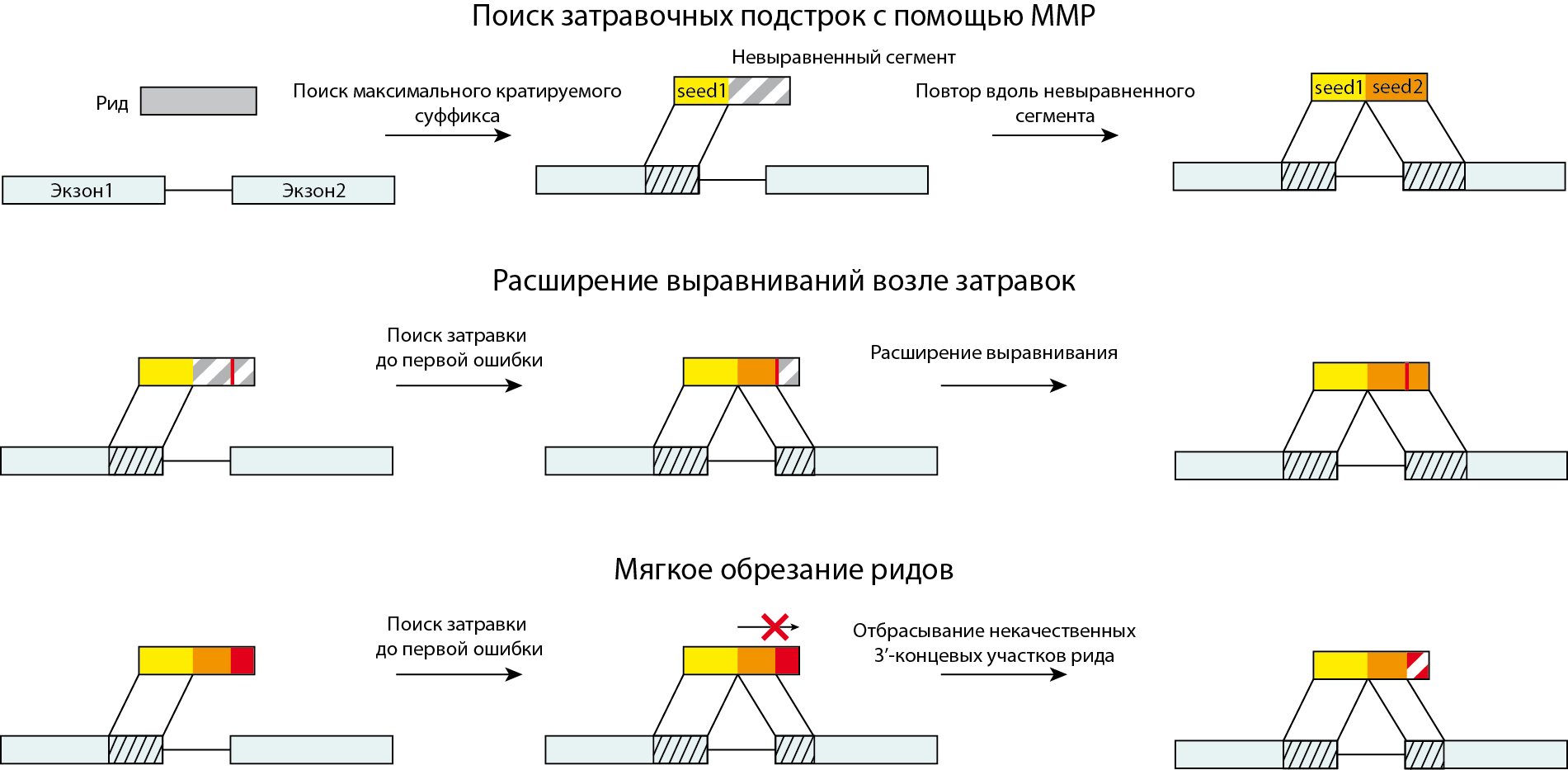
Большинство популярных инструментов выравнивания ридов RNA-seq были разработаны как усовершенствованные версии программ выравнивания непрерывных коротких последовательностей ДНК (bowtie, bwa и пр.) и основаны либо на выравнивания коротких прочтений по базе экзон-экзонных стыков (splice junctions), либо на выравнивании сегментов рида на непрерывную последовательность эталонного генома, либо на комбинации двух подходов. В отличие от них STAR был разработан для выравнивания сплайсированных последовательностей непосредственно на эталонный геномом. Алгоритм STAR состоит из двух основных этапов: этап поиска зерновых последовательностей (seed) и этап кластеризации/сшивки/скоринга (clustering/stitching/scoring).
Установка
Установка возможна только на Linux или OSX. Можно также попробовать запустить на Windows через Docker-клиент. Однако есть большая веротность, что ноутбук не справится с запуском программы из-за высоких требований к оперативной памяти.
Вариант 1. Клонирование репозитория из GutHub
В директории, где будет сохранена папка с программой, запустить команды:
git clone https://github.com/alexdobin/STAR.git
# Перейти в папку с исходными файлами программы
cd STAR/source
# Скомпилировать бинарные исходники
make STAR
cd ../bin
# Добавить путь к бинарным исходникам STAR в переменную PATH
export PATH=$PATH:$PWD
Вариант 2. Распаковка из архива
Копировать ссылку на скачивание .tar.gz архива с нужной версией программы на сайте GitHub (нажать правой кнопкой на гиперссылку “Source code (tar.gz)” в окошке с нужной версией программы и копировать).
Перейти в директорию, где будет храниться папка с программой STAR.
Вставить гиперссылку в команду wget, как показано ниже:
wget https://github.com/alexdobin/STAR/archive/2.7.10b.tar.gz
# скачается архив 2.7.10b.tar.gz
# распаковать его можно с помощью программы tar:
tar -xzf 2.7.10b.tar.gz
# появится папка STAR-2.7.10b. Можно сразу переименовать ее
mv STAR-2.7.10b STAR
# дальше делаем то же самое, вто и в Варианте 1:
cd STAR/source
# Скомпилировать бинарные исходники
make STAR
cd ../bin
# Добавить путь к бинарным исходникам STAR в переменную PATH
export PATH=$PATH:$PWD
Вариант 3. Самый простой. Установка при помощи Anaconda.
В командной строке вводим:
conda install -c bioconda star
Основные этапы выравнивания прочтений при помощи STAR
Как и в случае с другими элайнерами, выравнивание прочтений с помощью STAR — это двухэтапный процесс:
- Создание индекса на основе референсного генома.
Индекс - это сжатая версия текстовой последовательности генома. Фаловая структура индекса оптимизирована для быстрого поиска подпоследовательностей ДНК в геноме при помощи алгоритмов динамического программирования. Индекс создается один раз в папке назначения, и дальше используется неограниченное число раз для выравнивания ридов. Готовые индексы можно скачивать из интернета.
- Картирование прочтений на геном.
На этом этапе пользователь предоставляет папку с геномным индексом, а также файлы с прочтениями (последовательности) RNA-seq в формате FASTA или FASTQ. STAR сопоставляет прочтения с геномом и записывает несколько выходных файлов, включая выравнивание (SAM/BAM), сводную статистику, координаты экзон-экзонных стыков, невыравненные прочтения, профиль покрытия (wiggle) и т.д.
Дополнительные возможности STAR
В отличие от других популярных элайнеров STAR предназначен также для анализа single cell RNA-seq. Такая возможность реализована через алгоритм STARsolo, доступный в режиме soloCellFiltering, который помимо выравнивания прочтений на геном, собирает информацию о баркодах разного типа и подсчитывает экспрессию генов в единичных клетках.
Алгоритм STARconsensus, доступный начиная с версии STAR 2.7.7a, позоляет картировать риды на консенсусный геном, что может пригодиться для идентификации аллель-специфичных транскриптов. Для этого достаточно во время выравнивания вместе с референсным индексом выдать программе VCF-файл с геномными вариантами.
Помимо этого, в STAR заложены широкие возможности по поиску и аннотации сайтов сплайсинга. Программа также подходит для анализа химерных последовательностей ДНК, которые могут возникать из-за неканонических событий сплайсинга (включая транс-сплайсинг), геномных делеций/инверсий/инсерций или из-за ошибок обратной транскрипции/амплификации.
Один из режимов работы программы - создание профиля покрытия (signal file) генома прочтениями (inputAlignmentsFromBAM) в формате wiggle или BedGraph. Это может быть удобно для визуализации RNA-seq профиля в геномном браузере.
В режиме работы liftOver STAR конвертирует GTF-файл с аннотацией генома между геномными сборками (например, между dm3 и dm6). Для конвертации необходимо дать программе .chain файл, в котором сопоставляю координаты разных сборок.
Работа со STAR в командной строке
Запуск программы STAR происходит при помощи интерфейса командной строки. Программа запускается одной командой STAR, а режим работы (индексация генома, выравнивание и пр.) определяется обязательным параметром --runMode. Есть также версия программы STARlong, предназначенная, по-видимому, для выравнивания длинных прочтений. У нее практически те же самые параметры, что и у STAR (см. Полный
список параметров).
Индексация генома
Для создания индекса программе нужен:
- fasta-файл (или несколько файлов, указанных списком через пробел) с референсным геномом.
- [опционально] gtf-файл с координатами генов/транскриптов. STAR извлечет из этого файла координаты экзон-экзонных стыков (сплайс-сайтов) и использует их для значительного повышения точности картирования. Этот файл необязателен, и STAR можно запускать без аннотаций. В то же время, использование аннотаций настоятельно рекомендуется создателями программы всегда, когда они доступны. Начиная с версии 2.4.1a, аннотации также могут быть включены “на лету” на этапе картирования.
Индексированный геном включает двоичную последовательность генома, деревья суффиксов, текстовые названия/длины хромосом, координаты сплайс-сайтов и информацию о транскриптах/генах. Большинство этих файлов используют внутренний формат STAR и не предназначены для использования конечным пользователем. Настоятельно не рекомендуется изменять любой из этих файлов, за одним исключением: вы можете переименовать имена хромосом в файле chrName.txt, сохранив порядок хромосом в этом файле. Имена хромосом из этого файла будут использоваться во всех выходных файлах (например, SAM/BAM).
Параметры для индексации генома
Основные параметры для создания индексированного генома с использованием STAR следующие:
--runThreadN число_потоковустанавливает многопоточный режим--runMode genomeGenerateуказывает программе, что она должна создать индексированный геном--genomeDir /путь/к/папке/для/хранения/индексов--genomeFastaFiles /путь/к/FASTA1 /путь/к/FASTA2 ...--sjdbGTFfile /путь/к/GTF--sjdbOverhang длина_рида-1используется программой для построения базы аннотированных экзонных стыков. Если величина равна 100, то STAR будет индексировать последовательности длиной 100 пар с каждой стороны от сайта сплайсинга. В идеале эта величина должна составлять длину рида минус 1. Например, для запуска в режиме 2x150 bp (спаренные риды длиной 150 пар), идеальным значением будет 150-1=149. В случае, если риды имеют разную длину, то лучше выбрать длину самого протяженного рида и отнять единицу. Если этот параметр не указать, то программа воспользуется значением 100. Это значение отлично подходит в большинстве случаев.
По логике вещей, параметр –sjdbOverhang вообще может быть сколько угодно больше длины рида. Так что если вы индексировали геном с параметром 149, а реальная длина ридов 75, то на результат это никак не повлияет. Единственное, чем он больше, тем больше памяти занимает индекс. Вообще же, если вы не предоставляете gtf-файл с аннотацией генома, то на этот параметр можно не обращать внимания, ибо он ни на что не повлияет.
В итоге, типичный пример команды для индексации генома выглядит так (символ \ означает продолжение команды на новой строке):
STAR --runThreadN 12 \
--runMode genomeGenerate \
--genomeDir ath_star_index \
--genomeFastaFiles Athaliana_TAIR10.fasta \
--sjdbGTFfile Athaliana_gene.gtf \
--sjdbOverhang 149
Картирование прочтений
Для создания индекса программе нужен:
- Путь к папке с индексированным геномом.
- Пути к файлам с прочтениями в формате FASTA и FASTQ. Файлы могут быть сжатыми, однако STAR по умолчанию не умеет деархивировать файлы, поэтому в таком случае нужно написать команду, при помощи которой STAR сможет прочитать архивированные файлы.
- STAR также может извлекать риды из SAM/BAM-файлов, это может быть полезно для перевыравнивания.
- [опционально] Файл с аннотацией
Список файлов с прочтениями может быть задан при помощи табулированного manifest-файла.
Список файлов с прочтениями может быть задан при помощи табулированного manifest-файла.
Основные команды для картирования прочтений
Базовые параметры для картирования ридов перечислены ниже (в квадратных скобках показаны опциональные параметры):
--runRhreadN число_потоков
--genomeDir /путь/к/папке/для/хранения_индексов
--readFilesIn /путь/к/первому_риду [путь/ко/второму_риду]
В итоге, типичная команда выглядит следующим образом:
STAR --runThreadN 12 \
--readFilesIn ath_seed_sample.fastq \
--genomeDir ath_star_index
В результате в рабочей директории появится SAM-файл с выравненными прочтениями. Если понадобится выравнить парные прочтения, то команда выглядит так:
STAR --runThreadN 12 \
--readFilesIn ath_seed_sample_R1.fastq ath_seed_sample_R2.fastq \
--genomeDir ath_star_index
STAR не умеет работать со зжатыми файлами. Чтобы открыть их, ему необходимо написать в парметре --readFilesCommand команду, при помощи которой он сможет деархивировать файлы. Эта команда должна сгенерировать из входных файлов текст, который будет отправлен в STDOUT.
Для чтения файлов .fastq.gz, в этой опции нужно указать --readFilesCommand zcat или --readFilesCommand gunzip -c.
Для чтения файлов .fastq.bz, в этой опции нужно указать --readFilesCommand bzcat или --readFilesCommand bunzip2 -c.
Команда zcat или bzcat считывает сжатый текстовый файл и выводит в STDOUT, то есть на экран терминала. STAR использует текст из STDOUT как вводные данные.
Команды bunzip2 или gunzip деархивируют сжатые файлы и выводит содержимое в STDOUT при наличии опции -c.
Получается как-то так:
STAR --runThreadN 12 \
--readFilesIn ath_seed_sample_R1.fastq.gz ath_seed_sample_R2.fastq.gz \
--genomeDir ath_star_index \
--readFilesCommand zcat
Мы можем заставить STAR считывать риды не только из FATQ/FASTA, но также из SAM/BAM-файлов с выравниваниями. Это удобно, когда нужно перекартировать риды на другую сборку генома, для этого нужно указать тип исходного файла в опции --readFilesType как SAM SE (для односторонних прочтений) или как SAM PE (для двусторонних прочтений), а также заставить STAR запустить файл с помощью
samtools view (samtools должен быть установлен на устройстве). В таком случае команда будет выглядить так:
STAR --runThreadN 12 \
--readFilesIn aligned_SE.bam \
--readFilesType SAM SE \
--genomeDir ath_star_index \
--readFilesCommand samtools view
Обычно мы не работаем с неотсортированными SAM-файлами, а отдаем предпочтение отсортированным BAM-файлам. STAR может сразу выдать результат в таком виде, для этого нужно указать тип выводного файла с выравниванием:
STAR --runThreadN 12 \
--readFilesIn ath_seed_sample_R1.fastq.gz ath_seed_sample_R2.fastq.gz \
--genomeDir ath_star_index \
--readFilesCommand zcat \
--outSAMtype BAM SortedByCoordinate
Мы также можем заставить STAR сохранять файлы вывода в папку вне рабочей директории. В таком случае нужно попросить STAR дописывать к названиям файлов префикс, совпадающий с адресом папки назначения при помощи --outFileNamePrefix. Этот префикс должен содержать полный адрес директории назначения (заканчивается знаком /):
STAR --runThreadN 12 \
--readFilesIn ath_seed_sample_R1.fastq.gz ath_seed_sample_R2.fastq.gz \
--genomeDir ath_star_index \
--readFilesCommand zcat \
--outSAMtype BAM SortedByCoordinate \
--outFileNamePrefix /путь/к_папке/префикс_файла
В примере выше слово “префикс” находится после крайнего слэша / и будет первым словом в названии выводных файлов.
Также стоит отметить, что по умолчанию файл с выравниями не будет содержать риды, для которых выравнивания не обнаружилось (или которые не прошли фильтрацию). Чтобы изменить это поведение, нужно воспользоваться параметром --outSAMunmapped Within:
STAR --runThreadN 12 \
--readFilesIn ath_seed_sample_R1.fastq.gz ath_seed_sample_R2.fastq.gz \
--genomeDir ath_star_index \
--readFilesCommand zcat \
--outSAMtype BAM SortedByCoordinate \
--outFileNamePrefix /путь/к_папке/префикс_файла \
--outSAMunmapped Within
А если хочется вывести невыравненные прочтения в отдельный файл, то можно воспользоваться опцией --outReadsUnmapped Fastx, благодаря которой риды без выравнивания окажутся в файлах FASTQ/FASTA в зависимости от того, какой изначально тип файла с прочтениями был использован.
Фильтрация ридов с множественным картированием
Программа STAR обладает обширным списком настроек, контролирующих процесс фильтрации ридов с множественным картированием (когда рид одинаково хорошо выравнивается на несколько локусов генома). По этой причине STAR советуют использовать для анализа транскрипционной активности мобильных элементов.
Вывод ридов с множественным выравниванием (multimappers) контролируется через параметр
--outFilterMultimapNmax N. По умолчанию N=10. Если рид выравнивается на число локусов, меньшее или равное заданному то он появится в выравнивании. В ином случае он будет помечен как “Multimapping: mapped to too many loci” в лог-файле Log.final.out и будет считаться невыравненным.
Метод детекции ридов с множественным выравниванием контролируется параметром
--winAnchorMultimapNmax. По умолчанию здесь стоит значение 50. Этот параметр должен быть больше или равен --outFilterMultimapNmax. Однако от этого параметра зависит общая чувствительность выравнивания: чем он больше, тем дольше программа будет искать уникальные выравнивания.
Число локусов, на которое выравнился рид (Nmap) будет указано в атрибуте SAM-файла NH:i:Nmap. Очевидно, значение Nmap=1 будет соответствовать риду с уникальным картированием. Еще один атрибут HI будет использоваться для суммирования множественных вырваниваний рида, если их общее число превышает порог, указанный в параметре --outSAMattrIHstart. По умолчанию в этом параметре стоит значение 1,
однако в таких программах как Cufflinks требуется, чтобы порог был равен 0, для чего требуется ввести --outSAMattrIHstart 0.
В поле №5 (MAPQ) SAM-файла программа должна указать качество выравнивания. STAR автоматически ставит для ридов с уникальным выравниванием значение 255, а для ридов с множественным выравниванием int[-10*lg(1-1/Nmap)]. Однако для таких программ, таких как GATK, это значение потребуется изменить при помощи параметра --outSAMmapqUnique (принимает значение от 0 до 255).
В поле №2 (FLAG) SAM-файла программа должна указать, является ли выравнивание первичным (самым качественным из всех множественных выравниваний) или вторичным (вызывают сомнение, что рид пришел из этого локуса). Для ридов с множественным выравниванием все вырванивания кроме одного будут помечены флагом 0x100 (вторичное выравнивание). Одно случайное выравнивание с лучшей оценкой останенся
непомеченным и будет считаться первичным. Это поведение можно поменять при попощи опции --outSAMprimaryFlag AllBestScore, которая заставит программу пометить как первичные все выравнивания с наивысшей оценкой.
По умолчанию, порядок перечисления ридов с множественным выравниваниям не совсем случайный. Опция --outMultimapperOrder Random заставит выводить множественные выравнивания в случайном порядке и рандомизирует выбор первичного выравнивания из выборки с самой большой оценкой. Параметр --runRNGseed можно использовать для того, чтобы задать начальное значение (seed) генератора случайных чисел. При
одинаковом значении генератора результат выполнения программы будет максимально воспроизводимым. Нужно еще понимать, что использование многопоточного режима неизбежно приводит к отличиям в результатах.
Также может быть полезным ограничить число выводимых выравниваний для ридов с множественным картированием. Это ограничивается параметром --outSAMmultNmax. Однако даже если выводить только одно выравнивание, атрибут SAM-файла NH:i:Nmap все еще будет показывать точное количество локусов, на которое выравнился рид.
Результаты выполнения программы
В результате работы STAR выводит несколько файлов. По умолчанию все файлы имеют стандартные названия и будут загружаться в рабочую директорию. Чтобы изменить это поведение, необходимо передать программе префикс к названиям файлов. Это делается при помощи
--outFileNamePrefix /путь/к/директории/префикс_в_названии. Если хотите вывести все возможные выравнивания, передайте параметру --outSAMmultNmax значение -1.
Обратите внимение, префик содержит как путь к директории назначения, так и первые символы в названии файлов. Если хотите только поменять директорию назначения, то строка префикса должна заканчиваться прямым слэшем.
Логи
Программа выводит несколько log-файлов. Это обычные текстовые файлы, в которых содержится информация о запуске.
-
Log.out- это главный лог-файл, в котором перечислена основная информация о запуске программы. -
Log.progress.out- это файл, в котором перечисляется прогресс выполнения программы. Он обновляется раз в минуту. -
Log.final.out- это общая статистика по выравниваниям. Как и у других элайнеров, здесь перечисляется, сколько и каким образом выравнилось ридов. Нужно обратить внимание, что парные риды будут считаться за один. Большая часть информации приведена для ридов с уникальным вырваниванием. Уровень замен/инделов пересчитан на основе уникальных выравниваний и нормирован на общее число выравненных на геном оснований.
Файл с выравниваниями
Вы можете вывести файл с выравниваниями в формате SAM или BAM. В первом случае он будет называться Aligned.out.sam. Во втором - Aligned.out.bam.
Атрибуты в файле выравнивания
Помимо очевидных пунктов, таких как последователность рида, его локации в геноме, файлы выравниваний несут кучу полезной информации как о референсном геноме, так и об отдельных выравниваниях. Вся информация об атрибутах SAM или BAM приведена в спецификации SAM-файла.
STAR позволяет настроить выводимый список атрибутов при помощи опции --outSAMattributes. Полный перечень возможных атрибутов указан в разделе Настройки вывода SAM/BAM.
Совместимость с Cufflinks/Cuffdiff
Программы из семейства Cufflinks требуют, чтобы файлы выравниваний RNA-seq (если транскрипты секвенируются без сохранения цепи) содержали атрибут XS. Cufflinks требует, чтобы этот атрибут присутствовал у любого сплайсированного выравнивания. STAR генерирует этот атрибут с помощью опции --outSAMstrandField intronMotif.
Сплайсированные выравнивания, которые имеют неопределенную цепь (то есть неканонические экзон-экзонные стыки) в таком случае будут удалены (suppressed).
Если вы работаете с данными strand-specific RNA-seq, то в таком случае никаких дополнительных опций для STAR вводить не нужно - потребуется запустить Cufflinks с использованием параметра
--library-type.
Создатели STAR рекомендует также удалять риды, перекрывающие неканонические экзон-экзонные стыки, для запусков Cufflinks. Для этого нужно запустить STAR с параметром
--outFilterIntronMotifs RemoveNoncanonical.
Вывод файла с выравниваниями в формате BAM и сортировка
В работе часто мы не используем SAM-файлы. Вместо этого мы конвертируем их в BAM, поскольку так файлы занимают меньше места. Кроме того, большинство программ для анализа выравниваний требуют, чтобы выравнивания были отсортированы по координатам. Специально, чтобы пропустить шаг конвертации SAM в BAM, а также сортировку (обычно делается при помощи samtools sort), авторы STAR внедрили в свою
программу два этих шага.
Заставить STAR выводить несортированный BAM-файл можно при помощи опции --outSAMtype BAM Unsorted. В таком случае в директории появится файл Aligned.out.bam. Его можно дальше передать программе HTseq. Порядок ридов в таком несортированном файле будет точно соответсвовать порядку ридов в начальном файле FASTQ/FASTA.
Чтобы вывести сортированный BAM-файл, нужно ввести --outSAMtype BAM SortedByCoordinate. Тогда будет получен файл Aligned.sortedByCoord.out.bam. Это аналогично использованию samtools sort. Если сортировка таким образом вызывает ошибку, то рекомендуется понизить число потоков, которые используются при сортировке, при помощи --outBAMsortingThreadN (по умолчанию шесть потоков).
Если требуется вывести и сортированный и несортированный файл, то нужно ввести --outSAMtype BAM Unsorted SortedByCoordinate.
Вывод невыравненных прочтений
По умолчанию невыравненные риды (не удовлетворяющие параметрам фильтрации) будут проигнорированы. Если вы хотите вывести такие риды в основной файл с выравниваниями, то используйте опцию --outSAMunmapped Within. Если вы хотите вывести такие риды в отдельный файл с прочтениями (FASTQ/FASTA), то используйте --outReadsUnmapped Fastx.
Более подробно об этих опциях можно прочитать в разделе Настройки вывода SAM/BAM и Общие настройки вывода.
Сайты сплайсинга
STAR выводит информацию о достоверных сайтах сплайсинга в файл SJ.out.tab. Нужно обратить внимание, что точка разрыва сшивки программой STAR определяется по крайнему нуклеотиду в интроне.
Филтровать данные для этого файла можно при помощи параметра --outSJfilter. Подробности в разделе Фильтрация сплайс-сайтов.
Если хотите сохранить эту таблицу в файл с другим названием, можно восопльзоваться опцией --sjdbFileChrStartEnd /путь/к/сплайс_сайтам.tab
Для более чувствительного поиска сплайс-сайтов в STAR реализован режим двойного выравнивания (2-pass mode). Его можно включить при помощи опции --twopassMode Basic. В этом случае программа сначала выравнит заданное параметром twopass1readsN число ридов, чтобы предварительно подобрать сайты сплайсинга, а затем проведет 2й раунд (2nd pass) выравнивания, в котором уже все риды будут выравнены на
геном с учетом положения ранее выявленных сайтов сплайсинга. Если двойное выравнивание включено, то обнаруженные на первом этапе сайты сплайсинга (в доволнение к имеющимся в GTF-файле) будут считаться аннотированными (6 колонка в файле). Подробности режима двойного выравнивания можно прочитать в разделе Двухкратное картирование ридов для аннотации сайтов сплайсинга.
Вывод химерных выравниваний
STAR может детектировать химерные выравнивания в дополнению к обычным выравниваниям. Химерными считаются такие выравнивания, когда части ридов либо лежат слишком далеко, либо неправильно ложатся смысловую и антисмысловую часть ДНК (при каноническом сплайсинге спаренные риды должны быть на разных цепях ДНК, а части сплайсированного рида должны лежать на одной цепи ДНК), либо когда части ридов ложатся на разные хромосомы.
Создатели STAR определили химерные выравнинвания как выравнивания из двух нехимерных сегментов, причем сегменты должны лежать относительно друг друга как части химерной молекулы. Оба сегмента могут содержать сайты сплайсинга. Кроме того, один из сегментов может содержать фрагменты обоих спаренных ридов (т.е. риды перекрываются).
В последних версиях STAR поиск химерных выравниваний начинается, если разница оценки выравнивания у лучшего нехимерного выравнивания (с учетом мягкой обрезки) и лучшего сквозного выравнивания цельного рида больше чем число, указанное в --chimNonchimScoreDropMin (по умолчанию 20).
Парметр --chimSegmentMin контролирует минимальную длину двух сегментов. Например, для 2x75 прочтений, если --chimSegmentMin равен 20, то химерное выравнивание длиной 130 пар на одной хромосоме и 20 на другой хромосоме будет считаться химерным, тогда как выравнивание 135+15 не будет считаться химерным и будет удалено.
По умолчанию химерные выравнивания выводятся в табулированный файл Chimeric.out.junction.
Вы можете выводить химерные выравнивания в основной файл с выравниваниями, указав параметр --chimOutType WithinBAM..
Вы также можете вывести химерные выравнивания в отдельный SAM-файл --chimOutType SeparateSAMold - появится файл Chimeric.out.sam. Однако некоторые риды могут выводиться как в основной файл выравниваний, так и в химерный. Так происходит в тех случаях, если для рида есть два хороших выравнивания, причем одно из них химерное.
Программа STAR-Fusion
Создатели STAR также создали программу для детекции химерных транскриптов среди выравниваний STAR. Она доступна в репозитории GitHub.
Вывод выравниваний, попадающих внутрь генов
Используя опцию --quantMode TranscriptomeSAM, вы можете заставить STAR выводить только риды, которые выравниваются на аннотированные гены. В результате появится файл Aligned.toTranscriptome.out.bam. Это может быть полезно для вычисления экспрессии генов при помощи таких программ как RSEM или eXpress.
Алгоритм STAR в любом случае будет выравнивать риды на весь геном, а только после этого будет искать риды внутри генов. Пэтому в режиме --quantMode TranscriptomeSAM параметр --outFilterMultimapNmax (устанавливает максимальное число локусов, на которое может выравниваться рид) будет применен на стадии вырванивания на весь геном. Так что только те риды, которые проходят данный фильтр и попадают
в гены, будут выводиться.
Нужно иметь в виду, что по умолчанию в этом режиме STAR будет выводить риды, удовлетворяющие параметрам RSEM: мягкая обрезка или инделы запрещены. Чтобы изменить это поведение, нужно воспользоваться оцпией --quantTranscriptomeBan Singleend. Это может понадобиться для других программ, таких как eXpress.
Подсчет числа прочтений на ген
Одна из задач RNA-seq - количественная оценка активности генов. Это делается путем подсчета числа прочтений внутри каждого гена. Обычно для этого используются специальные программы, которые принимают файл выравниваний и файл с геномной аннотацией, чтобы посчитать число прочтений в заданных генах (FeatureCounts, HTseq-count и пр.). STAR разработан специально, чтобы избежать дополнительных упражнений с результатами и позволяет сразу вычислать число прочтений в генах.
Чтобы заставить STAR считать прочтения, нужно воспользоваться опцией --quantMode GeneCounts. Риды будут подсчитаны в том случае, если они пересекают ген (на 1 нуклеотид или более, как укажете). Оба конца спаренных ридов будут проверяться на предмет пересечения с геном. В результате вы получите файл с числом прочтений на ген, аналогичный результату работы HTseq-count в стандартном режиме.
Использование этой опции возможно только в том случае, если файл аннотации в формате GTF был добавлен г индесу или на стадии выравнивания при помощи опции --sjdbGEGfile.
В результате будет получен файл ReadsPerGene.out.tab.
Если поставить опцию --quantMode TranscriptomeSAM GeneCounts, то будут выведены файлы Aligned.toTranscriptome.out.bam и ReadsPerGene.out.tab.
2х-кратное картирование
Изначальная идея 2-хкратного картирования заключалается в следующем.
Для эксперимента, в котором будет проанализировано несколько образцов, советуют делать анализ в два этапа:
-
Выравнить каждый образец на геном, чтобы получить таблички с обнаруженными в ходе выравнивания сайтами сплайсинга.
-
Запустить выравнивание для каждого образца еще раз, только на этот раз перечислить аннотированные на первом этапе сайты сплайсинга с помощью
--sjdbFileChrStartEnd:--sjdbFileChrStartEnd /path/to/sj1.tab /path/to/sj2.tab ....
Чтобы не делать одну и ту же работу дважды, создатели STAR внедрили в программу возможность двойного картирования (2-pass mode), который включается при помощи опции --twopassMode Basic. Суть в том, что на первом этапе программа выравнивает заданное параметром twopass1readsN число ридов, чтобы предварительно подобрать неаннотированные сайты сплайсинга. На втором круге выравнивания (2nd pass), в
котором уже все риды будут выравнены на геном с учетом положения ранее выявленных интронов.
В более старых версиях STAR (до 2.4.1а) алгоритм 2х-кратного картирования отличался тем, что после первого прогона пользователю приходилось перезаписывать индексированный геном, добавляя в него новый файл с координатами экзон-экзонных стыков.
Слияние перекрывающихся спаренных ридов
Для того, чтобы увеличить точность картирования, в STAR реализована возможность автоматического слияния (merging) спаренных ридов. Это возможно, когда длина фрагмента ДНК, фланкированного секвенирующими адаптерами, оказывается меньше чем суммарная длина ридов. Например, в запуске 2x75 риды будут перекрываться для всех фрагментов короче 150 bp.
Чтобы заставить STAR сливать риды, нужно указать минимальную область пересечения в параметре --peOverlapNbasesMin и долю возможных замен в области перекрывания в --peOverlapMMp. Если перекрывание обнаружено, то STAR сливает два рида в одноцепочечную последовательность, которую будет пытаться выравнить на геном. Если такие слитые последовательности не выравниваются, то риды будут выравниваться
отдельно друг от друга.
Выравнивание с учетом мутантных вариантов
В некоторых случаях исследователям требуется вычислять экспрессию аллель-специфичных транскриптов. Один из вариантов такого анализа - добавить информацию об аллельном происхождении прочтения непосредственно в файл с выравниваниями.
Чтобы сделать это, вы можете добавить VCF-файл с перечисленными мутацианными вариантами на стадии выравнивания. Для этого нужно добавить адрес к VCF-файлу в опцию --varVCFfile /путь/к/vcf/файлу . В версии 2.7.10b возможен анализ только однонуклеотидных вариантов (SNV). Каждый SNV должен иметь два аллельных варинта. Варианты, которые
перекрываются с выравниванием, будут добавлены в атрибуты SAM-файла vG (геномная координата) и vA (собственно тип аллеля: 1 - аллель один, 2 - аллель два, 3 - ни один аллель не совпал). Нужно обязательно проконтролировать, чтобы эти атрибуты присутствовали в списке параметра --outSAMattributes.
Фильтрация аллель-специфичных выравниваний при помощи WASP
Чтобы дать пользователям возможность исследовать аллель-специфичную экспрессию, создатели добавили в STAR алгоритм [WASP] (https://www.nature.com/articles/nmeth.3582) для фильтрации аллель-специфичных прочтений. Она включается при помощи опции --waspOutputMode SAMtag.
После включения WASP в итоговом SAM-файле появится атрибут vW с несколькими возможными значениями: vW:i:1 - выравнивание прошло фильтрацию WASP vW:i:2 - рид имеет множество выравниваний vW:i:3 - вырожденный нуклеотид N (неизвестно, каким нуклеотидом представлен аллель) vW:i:4 - после повторного картирования рид не выравнивается vW:i:5 - после повторного картирования рид имеет несколько
выравниваний vW:i:6 - после повторного картирования рид выравнивается на другой локус vW:i:7 - рид перекрывает слишком много геномных варинатов
Выравнивание на консенсусный геном при помощи STARconsensus
В версии 2.7.7а создатели внедрили алгоритм STARconsensus, который предназначался для выравнивания ридов на индекс с учетом замен, предложенных в VCF-файле (consensus mapping). Это позволяло учесть как SNV, так и инделы.
Работает это следующим образом:
У вас на руках должен быть референсный геном (FASTA-файл), [опционално] файл с аннотированными генами или [опционално] таблица сплайс-сайтов, а также VCF-файл с перечисленными консенсусными геномными вариантами. Используя все это вы индексируете геном в режиме --runMode genomeGenerate. Чтобы добавить VCF-файл, нужно использовать параметр
--genomeTransformVC /путь/к/VCF, и указываете метод трансформации референса --genomeTransformType Haploid:
STAR --runThreadN 12 \
--runMode genomeGenerate \
--genomeDir /директория/для/будущего/индекса/ \
--genomeFastaFiles /путь/к/референсу.fa \
--sjdbGTFfile /файл/с/аннотацией.gtf \
--sjdbFileChrStartEnd /путь/к/сплайс_сайтам.tab \
--genomeTransformVC /путь/к/VCF \
--genomeTransformType Haploid \
--sjdbOverhang 149
Во время индексации STAR использует VCF-файл, чтобы модифицировать референс, и в результате получит трансформированный геном. Координаты генов тоже могут поменяться (из-за инделов), так что файл с аннотация тоже будет трансформирована.
На стадии картирования все риды будут выравнены уже на трансформированный геном. Вы можете посчитать экспрессию генов при помощи стандартной опции --quantMode TranscriptomeSAM и/или GeneCounts. Если нужно, то полученные выравнивания или сплайс-соединения (файл SJ.out.tab) могут быть трансформированы в обратно к координатам изначального референса при помощи --genomeTransformOutput SAM,
--genomeTransformOutput SJ или --genomeTransformOutput SAM SJ.
STARsolo для картирования, думультиплексирования и квантификации генов в single cell RNA-seq
С появлением технологий scRNA-seq, возникла необходимость в создании алгоритмов, специлизированных на обработки соответствующих транскриптомных данных. Разные платформы предлагают различные стратегии секвенирования транскриптома в масштабе единичных клеток, хотя общяя идея везде одна: добавить к секвенирующим адаптерам участки с вырожденной последовательностью (т.е. баркоды) таким образом, чтобы все прочтения, полученные из одной клетки, обладали уникальным баркодом. Помимо клеточных баркодов в адаптеры добавляют UMI - еще один баркод, уникальный для фрагмента ДНК. Если два рида имеют одинаковый UMI и клеточный баркод, то можно с уверенностью утверждать, что они являются ПЦР-дупликатами.
STARsolo представляет собой набор параметров, которые позволяют за один запуск программы решить несколько задач: выравнить риды на геном, определить принадлежность рида к клетке, определить среди ридов ПЦР дупликаты (провести демультиплексирование/дедупликацию при помощи UMI), посчитать экспрессию каждого гена в каждой клетке.
Из-за того, что секвенатор может совершать ошибки при прочтении нуклеотидов, а также из-за того, что ошибки могут возникать на стадии амплификации библиотек, STAR использует алгоритм коррекции ошибок (включает несколько параметров фильтрации), чтобы точно идентифицировать уникальные клеточные баркоды и UMI.
Алгоритм STARsolo заимствует много вещей у 10x CellRanger, так что может служить ествественной заменой этой программе.
Список опций для запуска STARsolo указан в разделе Параметры STARsolo (для анализа cingle cell RNA-seq), а подробное описание процедуры анализа scRNA-seq дано на странице создателей в GitHub
Конвертация GTF-файлов между геномными сборками
В инструкции к STAR указано, что в программе есть алгоритм конвертации собрки (liftOver). Подробного объяснения не дается, однако какието-вещи даны в описаниях к параметрам.
Для конвертации GTF-файла из сборки (например, dm3 в сборку dm6), требуется chain-файл, в котором приведено попарное вырванивание координат в разных геномных сборках. Скачать chain-файлы можно, например, с ftp-репозитория UCSC. Обратите внимание, что ссылка на chain-файлы (LiftOver files) здесь
дана отдельно для каждого организма. Кроме того, chain-файлы будут отличаться в зависимости от направления конвертации. Напремер, для конвертации dm3 в dm6 нужен файл dm3ToDm6.over.chain.gz, а при обратной конвертации dm6ToDm3.over.chain.gz.
Далее у нас есть файл аннотации GTF для dm3, и чтобы перевести его в dm6 нужно ввести следующее:
STAR --runMode liftOver \
--sjdbGTFfile /путь/к/dm3.gtf
--genomeChainFiles /путь/k/dm3ToDm6.over.chain.gz
--readFilesCommand zcat
Обратите внимание, так как STAR не умеет читать сжатые файлы, скорее всего придется воспользоваться опцией –readFilesCommand. По идее, в рабочей директории будет создан файл GTF, конвертированный под сборку dm6.
Создание профиля покрытия из выравниваний
Также в описании к программе STAR есть пункты о возможности создать профиль покрытия (signal file, аналог DeepTools bamCoverage). Информации об этом мало, какая-то информация дана в оригинальной статье про STAR, но судя по всему нужно сделать следующее:
STAR --runMode inputAlignmentsFromBAM \
--outWigType wiggle | bedGraph | bedGraph read1_5p | bedGraph read2 \
--readFilesIn /путь/к/bam_или_sam-файлу \
[--outWigStrand Stranded |Unstranded] \
[--readFilesCommand samtools view] \
[--outWigNorm RPM | None]
Здесь альтернативные значения параметров указаны через прямую черту |. Необязательные параметры (но я не уверен, что они необязательны) указаны в квадратных скобках.
Подробнее о параметрах профиля покрытия можно узнать в разделе Вывод профиля покрытия в формате Wiggle или BedGraph.
Полный список параметров
Список параметров приведен в соответсвии с версией программы 2.7.10b.
Файл с параметрами
| Параметр | Значение по умолчанию | Описание |
|---|---|---|
--parametersFiles |
- |
путь к файлу Путь к файлу со списком заданных параметров для запуска в командной строке. “-” - путь по умолчанию, соответсвует
файлу |
Выбор системной оболочки
| Параметр | Значение по умолчанию | Описание |
|---|---|---|
--sysShell |
- |
путь к бинарному файлу Путь к интерпретатору оболочки в Unix-системе. “-” - путь по умолчанию в системе (обычно |
Режим работы программы
| Параметр | Значение по умолчанию | Описание |
|---|---|---|
--runMode |
alignReads |
строка Определяет режим работы программы:
|
--runThreadN |
1 |
целое число Число потоков |
--runDirPerm |
User_RWX |
строка Разрешения на запись, чтение, удаление файлов во время работы программы
|
--runRNGseed |
777 |
целое число Зерно для генератора случайных чисел. Два одинаковых запуска программы с разным зерном дадут немного различающиеся результаты. |
Параметры индексированного генома
| Параметр | Значение по умолчанию | Описание |
|---|---|---|
--genomeDir |
./GenomeDir/ |
путь к директории Путь к директории с индексированным геномом. В режиме |
--genomeLoad |
NoSharedMemory |
строка Порядок действий с разделяемой
памятью
при использовании файлов генома. Разделяемая память позволяет нескольким запущенным программам STAR обращаться к одному
разделу памяти, сократив время запуска программы. Используется только в режиме
|
--genomeFastaFiles |
- |
адрес файла Путь к FASTA-файлам с референсными последовательностями генома. Файлы не могут быть сжатыми. Обязательно указывается в
режиме |
--genomeChainFiles |
- |
адрес файла Путь к .chain файлу для
конвертации (lift-over) GTF между версиями сборки
генома. Используется только в режиме |
--genomeFileSizes |
0 |
целое число Точные размеры индексов в байтах. Не используется юзерами. |
--genomeTransformOutput |
None |
строка Используется при картировании RNA-seq ридов на консенсусный геном алгоритмом STARconsensus, когда результирующие координаты выравниваний или экзонных-стыков сдвигаются. При необходимости можно заставить программу трансформировать результирующие координаты в исходные, соотвествующие изначальному референсу. Варианты: |
--genomeChrSetMitochondrial |
chrM M MT |
строка Названия митохондриальных хромосом. Испольуется только для вывода статистики алгоритмом STARsolo. |
Параметры индексирования генома в режиме genomeGenerate
| Параметр | Значение по умолчанию | Описание |
|---|---|---|
--genomeChrBinNbits |
18 |
целое число При хранении генома каждая хромосома будет занимать целое число бинов. Параметр определяет размер бина через показательную функцию log2(chrBin). При работе с геномами, в которых большое число контигов, этот параметр нужно снизить. Если обозначить длину генома за L, число последовательностей в референсном геноме за N, а длину рида за l, то наилучшим значением для этого параметра будет min(18, log2[max(L/N,l)]). |
--genomeSAindexNbases |
14 |
целое число Длина преиндексированной строки в дереве суффиксов (SA). Обычно между 10 и 15. Более длинные строки требуют больше памяти, но ускоряют поиск. Для маленьких геномов (меньше 1Gb) должнобыть уменьшено согласно формуле: min(14, log2(L)/2 - 1), где L - длина генома. |
--genomeSAsparseD |
1 |
целое положительное число Разряженность (sparsity) дерева суффиксов, ака дистанция между индексированными суффиксами. Чем больше число, тем меньше требования к оперативной памати, но медленнее выравнивание. |
--genomeSuffixLengthMax |
-1 |
целое число максимальная длина суффиксов. Должна быть больше длины ридов. Значение |
--genomeTransformType |
None |
строка Тип трансформации генома:
|
--genomeTransformVCF |
- |
путь к файлу Путь к VCF-файлу для трансформации генома |
Параметры базы экзонных стыков (Splice Junctions)
| Параметр | Значение по умолчанию | Описание |
|---|---|---|
--sjdbFileChrStartEnd |
- |
путь к файлу Путь к файлу с геномными координатами экзон-экзонных стыков. Может быть несколько файлов, перечисленных через пробел. |
--sjdbGTFfile |
- |
путь к файлу Путь к GTF-файлу с аннотацией генома. |
--sjdbGTFchrPrefix |
- |
строка префикс в начале имени хромосомы в GTF-файле. Как правило это “chr”. Может пригодиться при использовании аннотации ENSEMBL и референса UCSC. |
--sjdbGTFfeatureExon |
exon |
строка Тип объекта (feature) в GTF-файле, который будет использоваться в качестве экзонов при построении транскриптов. |
--sjdbGTFtagExonParentTranscript |
transcript_id |
строка Название атрибута в GTF-файле, который будет использоваться качестве идентификатора (ID) порождающего транскрипта (parent transcript). |
--sjdbGTFtagExonParentGene |
gene_id |
строка Название атрибута в GTF-файле, который будет использоваться качестве идентификатора (ID) порождающего гена (parent gene). |
--sjdbGTFtagExonParentGeneName |
gene_name |
строка Название атрибута в GTF-файле, который будет использоваться качестве названия (name) порождающего гена (parent gene). |
--sjdbGTFtagExonParentGeneType |
gene_type gene_biotype |
строка Название атрибутов в GTF-файле, который будет использоваться качестве типа (белок-кодирующий, некодирующий и пр.) порождающего гена (parent gene type). |
--sjdbOverhang |
100 |
целое положительное число длина донорской/акцепторной последовательности с каждой стороны экзон-экзонного стыка. В идеале должно быть на 1 короче длины рида. |
--sjdbScore |
2 |
целое число Дополнительный бонус за выравнивание (extra alignment score), которое пересекает базу экзонных стыков. |
--sjdbInsertSave |
Basic |
строка Указывает, какие файлы сохранять, когда информация об экзонных стыках добавляется на стадии выравнивания:
|
Добавление геномных вариантов
| Параметр | Значение по умолчанию | Описание |
|---|---|---|
--varVCFfile |
- |
путь к файлу Путь к файлу VCF c данными по вариации генома. В 10й колонке должен содержать информацию о генотипе в формате 0/1. |
Вводные файлы для профиля покрытия
| Параметр | Значение по умолчанию | Описание |
|---|---|---|
--inputBAMfile |
- |
путь к файлу Путь к файлу BAM-файлу для построения профилей покрытия генома в режиме |
Параметры базы экзонных стыков (Splice Junctions)
| Параметр | Значение по умолчанию | Описание |
|---|---|---|
--sjdbFileChrStartEnd |
- |
путь к файлу Путь к файлу с геномными координатами экзон-экзонных стыков. Может быть несколько файлов, перечисленных через пробел. |
--sjdbGTFfile |
- |
путь к файлу Путь к GTF-файлу с аннотацией генома. |
--sjdbGTFchrPrefix |
- |
строка префикс в начале имени хромосомы в GTF-файле. Как правило это “chr”. Может пригодиться при использовании аннотации ENSEMBL и референса UCSC. |
--sjdbGTFfeatureExon |
exon |
строка Название атрибута в GTF-файле, который будет использоваться в качестве экзонов при построении транскриптов. |
--sjdbGTFtagExonParentTranscript |
transcript_id |
строка Название атрибута в GTF-файле, который будет использоваться качестве идентификатора (ID) порождающего транскрипта (parent transcript). |
--sjdbGTFtagExonParentGene |
gene_id |
строка Название атрибута в GTF-файле, который будет использоваться качестве идентификатора (ID) порождающего гена (parent gene). |
--sjdbGTFtagExonParentGeneName |
gene_name |
строка Название атрибута в GTF-файле, который будет использоваться качестве названия (name) порождающего гена (parent gene). |
--sjdbGTFtagExonParentGeneType |
gene_type gene_biotype |
строка Название атрибутов в GTF-файле, который будет использоваться качестве типа (белок-кодирующий, некодирующий и пр.) порождающего гена (parent gene type). |
--sjdbOverhang |
100 |
целое положительное число длина д онорской/акцепторной последовательности с каждой стороны экзон-экзонного стыка. В идеале должно быть на 1 короче длины рида. |
--sjdbScore |
2 |
целое число Дополнительный бонус за выравнивание (extra alignment score), которое пересекает базу экзонных стыков. |
--sjdbInsertSave |
Basic |
строка Указывает, какие файлы сохранять, когда информация об экзонных стыках добавляется на стадии выравнивания:
|
Параметры прочтений
| Параметр | Значение по умолчанию | Описание |
|---|---|---|
--readFilesType |
Fastx |
строка Формат, в котором предоставлены риды:
1 - При работе с BAM файлами используйте параметр |
--readFilesSAMattrKeep |
All |
строка Указывает, какие флаги в SAM-файле, предоставленном в качестве файла с прочтениями
|
--readFilesIn |
Read1 Read2 |
пути к файлам Адреса файлов c прочтениями. Сначала первый рид (или список через запятую), потом спаренные риды через пробел (если есть). Примеры:
Если файлов с ридами много (указаны через запятые), то в файл выравнивания можно добавить специальный атрибут
В примере выравниваются три файла, риды из каждого будут помечены. Обратите внимание, префикс |
--readFilesManifest |
- |
путь к файлу Путь к файлу с таблицей, содержащей адреса файлов с прочтениями (manifest file). Это дает возможность выравнивать несколько экспериментов в одном запуске. Таблица состоит из трех столбцов, разделенных табуляцией: file1_R1.fq (tab) file1_R2.fq (tab) строка_флагов1 file2_R1.fq (tab) file2_R2.fq (tab) строка_флагов2 Для случая с односторонними прочтениями вместо второго рида указывается file1.fq (tab) - (tab) строка_флагов1 file2.fq (tab) - (tab) строка_флагов2 Строка флагов может начинаться с префикса Если в строке флагов нет префикса Все флаги будут записаны дословно в строку заголовков |
--readFilesCommand |
- |
строка Команда, которую STAR должен запустить в командной строке перед загрузкой файлов с прочтениями. STAR не умеет работать со зжатыми файлами. Чтобы открыть их, ему необходимо написать команду, при помощи которой он сможет деархивировать файлы. Эта команда должна сгенерировать из входных файлов текст, который будет
отправлен в Для чтения файлов Для чтения файлов 1 - команда 2 - команды |
--readFilesPrefix |
- |
строка Префикс, который нужно добавить к названиям файлов в команде |
--readMapNumber |
-1 |
целое число Число ридов с начала файла, которые нужно выравнить на геном. Позволяет делать проверочные запуски выравнивания. Значение |
--readMatesLengthsIn |
NotEqual |
строка Стоит ли программе ожидать, что длина имен, последовательностей, строки качества в файлах со спаренными ридами одинакова. Варианты: |
--readNameSeparator |
/ |
строка Символ в названии ридов (в FASTQ-файлах это строка, начинающаяся с |
--readQualityScoreBase |
33 |
неотрицательное целое число Базовое значение для кодировки Phred -score. Символ с этим номером в таблице ASCII будет восприниматься как Phred=0. Для запусков Illumina использовать 33. Для запусков Solexa - 64. |
Обрезака ридов
| Параметр | Значение по умолчанию | Описание |
|---|---|---|
--clipAdapterType |
Hamming |
строка Способ удаления адаптера:
|
--clip3pNbases |
0 |
целые числа Число оснований, которое будет удалено с 3’ конца каждой пары. Если задано одно число, то программа будет удалять заданное количетсво букв у обоих спаренных ридов. |
--clip3pAdapterSeq |
- |
строка Удалит заданную последовательность адаптера с 3’ конца ридов. Если записана одна последовательность, то левый и правый риды будут обризаться одинаково. Можно задать значение |
--clip3pAdapterMMp |
0.1 |
вечественное число максимальная доля несовпадающих букв на 3’-конце рида в сравнении с последовательностью адаптера. Если приведено два числа, то для левого и правого рида этот параметр будет отличаться. |
--clip3pAfterAdapterNbases |
0 |
целое число Число оснований, которое будет удалено с 3’-конца ридов после удаления адаптеров. Можно указать два числа для разных спаренных ридов. |
--clip5pNbases |
0 |
целое число Число оснований, которое будет удалено с 5’ конца каждой пары. Если задано одно число, то программа будет удалять заданное количетсво букв у обоих спаренных ридов. |
Ограничения памяти
| Параметр | Значение по умолчанию | Описание |
|---|---|---|
--limitGenomeGenerateRAM |
31000000000 |
целое положительное число Максимальный объем оперативной памяти (байт) для генерации генома |
--limitIObufferSize |
30000000 50000000 |
целое положительное число Максимальный объем буфера (байт) для ввода/вывода на поток |
--limitOutSAMoneReadBytes |
100000 |
целое положительное число Максимальный размер одной записи в SAM-файле (байт) на рид. Рекомендуется значение: > (2*(LengthMate1+LengthMate2 +100) * outFilterMultimapNmax |
--limitOutSJoneRead |
1000 |
целое положительное число Максимальное число экзонных стыков на рид (учитывая все множественные выравнивания) |
--limitOutSJcollapsed |
1000000 |
целое положительное число Максимальное число экзонных стыков после удаления дупликатов |
--limitBAMsortRAM |
0 |
целое положительное число Максимальное количество оперативной памяти для сортировки BAM. Если равно 0, то будет равно размеру индекса. Значение 0 может быть использовано только вместе с параметром |
--limitSjdbInsertNsj |
1000000 |
целое положительное число Максимальное число экзонных стыков, которое можно вставить в том случае, если анализ экзон-экзонных стыков происходит на стадии выравнивания. |
--limitNreadsSoft |
-1 |
целое число Мягкое ограничение на число ридов. |
Общие настройки вывода
| Параметр | Значение по умолчанию | Описание |
|---|---|---|
--outFileNamePrefix |
./ |
строка Префикс, который будет добавлен к названию файлов оутпута. Это может быть адрес папки, куда вы хотите сохранить все файлы. |
--outTmpDir |
- |
строка Fastxпуть к директории для временных файлов. Все содержимое будет удалено по покончании программы. |
--outTmpKeep |
None |
строка Сохранять временные файлы после завершения программы (выбрать |
--outStd |
Log |
строка Указывает, какой из файлов будет направлен в STDOUT (на экран терминала). Варианты - логи |
--outReadsUnmapped |
None |
строка Указывает, сохранить ли невыравненные или частичновыравненные риды в отдельный файл. Есть вариант |
--outQSconversionAdd |
0 |
целое число Заставляет добавить число к Phred-score. Позволяет конвертировать Phred между кодировкой +33 и +64. Например, можно добавить к значениям качества Illumina 31, чтобы получить кодировку Phred+64. |
--outMultimapperOrder |
Old_2.4 |
строка Порядок перечисления ридов с множественными выравниваниями в результатах:
|
Настройки вывода SAM/BAM
| Параметр | Значение по умолчанию | Описание |
|---|---|---|
--outSAMtype |
SAM |
строка Тип вывода SAM/BAM. Параметр принимает одно или два слова. Первое означает тип файла выравнивания: Второе (опционально):
|
--outSAMmode |
Full |
строка Метод вывода в SAM:
|
--outSAMstrandField |
None |
строка Заставляет программу записывать в SAM-файл информацию о цепочке ДНК специально, чтобы результат воспринимался программой Cufflinks. Это необходимо для совместимости между файлом выравнивания и некоторыми программами анализа.
|
--outSAMattributes |
Standard |
строка Задает список атрибутов SAM-файла, которые нужно вывести. Эти параметры будут находиться в 12-м опциональном поле таблицы вырвавниваний в виде записей формата Тэг:Tип_ переменной:Значение. Например,
Пресеты:
Варианты атрибутов, описывающих вырванивание:
Варианты атрибутов, доступные в режиме работы с мутантными вариантами:
Варианты атрибутов, доступные в режиме работы STARsolo (д емультиплексирование scRNA-seq выравниваний):
Кастомные атрибуты, отсутствуют в официальной спецификации SAM/BAM:
|
--outSAMattrIHstart |
1 |
целое неотрицательное число Начальное значение для атрибута IH. Значение 0 может быть обязательным для некоторых программ, таких как Cufflinks или StringTie |
--outSAMunmapped |
None |
строка Указывает, как выводить некартированные риды. Параметр состоит из двух частей. Первая часть:
Вторая часть:
|
--outSAMorder |
Paired |
строка Тип сортировка для ридов в выравнивании:
|
--outSAMprimaryFlag |
OneBestScore |
строка Указывает, какое выравнивание считать первичным. Все остальные выравнивания получат флаг 0x100 (В флаге восьмой бит справа будет приравнен 0).
FLAG - это двоичное 16-битовое число, каждая цифра в котором указывает на определенное свойство рида. Например, если первый бит (0x1 в 16-ричной нотации) равен единице, то рид считается спаренным (0 - наоборот). Если второй бит (0x2 в 16-ричной нотации) равен 1, то рид выравнен правильно вместе со своей парой (0 - наоборот). Если записать два бита одним двоичным числом, то число 112 означает, что рид имеет пару, и оба рида в паре правильно выравнены. А если мы видим число 012, то рид имеет пару, но оба выранены неправильно (дискордантно, например). Любое число в двоичной системе можно выразить в десятеричной. Например, 112 эквивалентно 410. Расшифровку флагов можно посмотреть в онлайн-калькуляторе, его более красивой версии, или видео туториале. |
--outSAMreadID |
Standard |
строка Тип записи для идентификатора рида (read ID):
Read ID указан в первой колонке SAM-файла (поле QNAME). |
--outSAMmapqUnique |
255 |
целое число от 0 до 255 Устанавливает качество выравнивания для у никально-картируемых ридов. Это число будет записано во второй колонке (поле MAPQ) внутри SAM-файла. |
--outSAMflagOR |
0 |
целое число от 0 до 65535 Изменяет флаги в SAM-файле при помощи операции OR. После того, как STAR сформирует выравнивания, раздел FLAG SAM-файла будет сложет с заданным числом. Может пригодится для фильтрации выравниваний на основе флагов. Подробнее про флаги можно прочитать в разделе про Поскольку в двоичной форме с любой цифрой можно проводить логические операции, то флаги можно модифицировать. STAR позволяет конвертировать флаги в итоговом выравнивании с помощью операций AND и OR. Например, можно модифицировать флаг 00 с помощью операции 01 AND 10. Получится 00. Или можно выполнить операцию 01 OR 10 - получится 11. |
--outSAMflagAND |
65535 |
целое число от 0 до 65535 Изменяет флаги в SAM-файле при помощи операции AND. После того, как STAR сформирует выравнивания, раздел FLAG SAM-файла будет сложет с заданным числом. Может пригодится для фильтрации выравниваний на основе флагов. Подробнее про флаги можно прочитать в разделе про Подробнее про операции над флагами можно прочитать в разделе про |
--outSAMattrRGline |
- |
строка Задает строку атрибутов в выравниваниях SAM-файла. Должна начинаться с тега Атрибуты для разных вводных файлов могут отличаться, для чего нужно будет написать несколько строк атрибутов через пр обел-запятую-пробел:
Обратите внимание, что теги, чье содержимое содержит пробелы, нужно писать в кавычках. |
--outSAMheaderHD |
- |
строка Задает первую строку заголовка (несет @HD флаг) SAM |
--outSAMheaderPG |
- |
строка Дополнительная строка в SAM-заголовке. Будет нести тег @PG (программное обеспечение). |
--outSAMheaderCommentFile |
- |
путь к файлу название файла, куда буду записаны коммендарии из SAM заголовка (несет тег @CO) |
--outSAMfilter |
None |
строка Указывает, нужно ли записывать выравнивания на референсные последовательности, добавленные с помощью
По умолчанию стоит |
--outSAMmultNmax |
-1 |
целое Максимальное число выравниваний, которое сообщается для ридов с множественными выравниваниями. По умолчанию стоит -1, то есть сообщаются все (влоть до |
--outSAMtlen |
1 |
целое метод вычисления поля TLEN (template length, длина фрагмента) в SAM/BAM файле.
|
--outBAMcompression |
-1 |
целое от -1 до 10 Уровень сжатия BAM-файла. Чем больше, тем сильнее сжатие. -1 означает обычное сжатие (~6). |
--outBAMsortingThreadN |
0 |
целое неотричательное число Число потоков для сортировки BAM-файла |
--outBAMsortingBinsN |
50 |
целое положительное число Число геномных фрагментов при сортировке по координатам |
Процессинг BAM-файлов
| Параметр | Значение по умолчанию | Описание |
|---|---|---|
--bamRemoveDuplicatesTpe |
- |
строка Устанавливать метки на ПЦР-дупликаты. В версии 2.7.10b опция работает только для сортированных BAM-файлов и только для парных прочтений.
|
--bamRemoveDuplicatesMate2basesN |
0 |
целое положительное число Устанавливает число основани с 5’конца рида R2, которое можно использовать для слияния ридов (используется для идентификации TSS в RAMPAGE). |
Вывод профиля покрытия в формате Wiggle или BedGraph
| Параметр | Значение по умолчанию | Описание |
|---|---|---|
--outWigType |
None |
строка Указывает тип файла покрытия:
Величину сигнала внутри бина генома можно считать по фрагменту, 5’ концу первого рида (read1_5p) и по второму риду (read2). Последние варианты полезны для CAGE или RAMPAGE. |
--outWigStrand |
Stranded |
строка
|
--outWigReferencesPrefix |
- |
строка префикс в названии хромосом, который нужно вставить в wig-файл. Как правило, это “chr”. Может понадобиться для отображения wig-файла в UCSC-браузере. Значение |
--outWigNorm |
RPM |
строка Метод нормализации числа прочтений на бин: |
Фильтрация выравниваний
| Параметр | Значение по умолчанию | Описание |
|---|---|---|
--outFilterType |
Normal |
строка Тип фильтрации выравненных прочтений.
|
--outFilterMultimapScoreRange |
1 |
целое число Оценка выравнивания максимального скора для фильтрации множественных выравниваний. |
--outFilterMultimapNmax |
10 |
строка Максимальное число локусов, на которое может выравниваться рид. Если локусов выравнивания больше, то ни одно из выравниваний не
сообщается, а рид будет помечен как “mapped to too many loci” в файле |
--outFilterMismatchNmax |
10 |
вещественное число Метод нормализации числа прочтений на бин: |
--outFilterMismatchNoverLmax |
0.3 |
вещественное число Выравнивание будет сообщаться только в том случае, если оотношение числа ошибочных оснований к точно совпавшим с референсом будет меньше или равно заданному числу. |
--outFilterMismatchNoverReadLmax |
1.0 |
вещественное число Выравнивание будет сообщаться только в том случае, если оотношение числа ошибочных оснований к длине рида меньше или равно заданному значению. |
--outFilterScoreMin |
0 |
целое Выравнивание будет сообщаться только в случае, если оценка за вырвавнивание не ниже заданного числа. |
--outFilterScoreMinOverLread |
0.66 |
вещественное число Тот же параметр, что и |
--outFilterMatchNmin |
0 |
целое Выравнивание будет сообщаться только в случае, если число совпавших с референсом основание превышает указанное. |
--outFilterMatchNminOverLread |
0.66 |
вещественное число Тот же параметр, что и |
--outFilterIntronMotifs |
None |
строка Фильтрация сплайс-сайтов.
|
--outFilterIntronStrands |
RemoveInconsistentStrands |
строка Фильрация сплайс-сайтов по направлению ДНК
|
Вывод сплайс сайтов в SJ.out.tab
| Параметр | Значение по умолчанию | Описание |
|---|---|---|
--outSJtype |
Standard |
строка Указывает, выводить ли идентифицированные экзонные стыки в |
Фильтрация сплайс-сайтов
| Параметр | Значение по умолчанию | Описание |
|---|---|---|
--outSJfilterReads |
All |
Указывает, какие риды добавлять в список сплайс-соединени:
|
--outSJfilterOverhangMin |
30 12 12 12 |
четыре целых числа Минимальная длина оверхэнга в неаннотированных сплайс-соединениях (с обеих сторон). Каждое число определяет разные типы мотивов на стыке сплайс-соединений:
Значение |
--outSJfilterCountUniqueMin |
3 1 1 1 |
четыре целых числа Минимальная число уникально картируемых ридов на неаннотированных сайт сплайсинга. Каждое число определяет разные типы мотивов на стыке сплайс-соединений:
Значение Сайт сплайсинга будет считаться валидным, если один из параметров |
--outSJfilterCountTotalMin |
3 1 1 1 |
четыре целых числа Аналогично |
--outSJfilterDistToOtherSJmin |
10 0 5 10 |
четыре целых неотрицательных числа минимально допустипая дистанция до следующего донор/акцепторного сайта сплайсинга. Не применяется к аннотированным сайтам сплайсинга. |
--outSJfilterIntronMaxVsReadN |
50000 100000 200000 |
N целых неотрицательных чисел Максимальная длина интрона (длина промежутка между донорским и акцепторным сайтом), поддержанного 1,2,3… N выравненными ридами. Чтобы сплайс-сайт, обрамляющий интрон длиной 50000bp считался валидным, нужен всего 1 выравненный на сплайс-соединение рид. Чтобы сплайс-сайт, обрамляющий интрон длиной 100000bp считался валидным, нужно уже два рида. Для интрона 200000bp, число выравненных на сплайс-соединение ридов должно составлять 3 шт. и т.д. |
Оценка выравниваний
| Параметр | Значение по умолчанию | Описание |
|---|---|---|
--scoreGap |
0 |
целое штраф (пенальти) за сплайс-сайт |
--scoreGapNoncan |
-8 |
целое штраф за неканоническое сплайс-соединение (добавляется к |
--scoreGapGCAG |
-4 |
целое пенальти за GC/AG и CT/GC сплайс-соединение (добавляется к |
--scoreGapATAC |
-8 |
целое пенальти за AT/AC и GT/AT сплайс-соединение (добавляется к |
--scoreGenomicLengthLog2scale |
-0.25 |
вещественное число Дополнительный бонус, логарифмически нормированный на геномную длину выравнивания: scoreGenomicLengthLog2scale* log2(genomicLength) |
--scoreDelOpen |
-2 |
целое Штраф за открытие делеции. Если в делеции (пропуск в риде относительно референса) три нуклеотида, то штраф за открытие дается за первый нуклеотид. За каждый дополнительный нуклеотид добавляется штраф за продолжение делеции. |
--scoreDelBase |
-2 |
целое Штраф за продолжение делеции. Добавляется за каждый нуклеотид в делеции кроме первого, суммируется с Пояснение смотреть в разделе про |
--scoreInsOpen |
-2 |
целое Штрав за начало встройки. Если во встройке (инсерция нуклеотидов в риде) три нуклеотида, то штраф за открытие дается за первый нуклеотид. За каждый дополнительный нуклеотид добавляется штраф за продолжение встройки. |
--scoreInsBase |
-2 |
целое Штраф за продолжение встройки. Добавляется за каждый нуклеотид в делеции кроме первого, суммируется с Пояснение смотреть в разделе про |
--scoreStitchSJshift |
1 |
целое Максимальное снижение оценки за выравнивание в процессе поиска сплайс-сайтов на этапе сшивки. |
Параметры выравнивания и поиска затравочной подстроки
| Параметр | Значение по умолчанию | Описание |
|---|---|---|
--seedSearchStartLmax |
50 |
целое положительное число Определяет точку начала поиска внутри рида. Рид разделяется на фрагменты не короче заданного числа. |
--seedSearchStartLmaxOverLread |
1.0 |
вещественное неотрицательное число То же, что и |
--seedSearchLmax |
0 |
целое неотрицательное число Определяет максимальную длину затравочной подстроки. Если 0, то ограничения нет. |
--seedMultimapNmax |
10000 |
целое положительное число Только куски ридов, которые картируются меньше указанного числа раз, участвуют в процедуре сшивания. |
--seedPerReadNmax |
1000 |
целое положительное число Максимальное число затравок на рид. |
--seedPerWindowNmax |
50 |
целое положительное число Максимальное число затравок на окно. |
--seedNoneLociPerWindow |
10 |
целое положительное число Максимальное число локусов с одним зерном на окно выравнивания. |
--seedSplitMin |
12 |
целое положительное число Минимальная длина затравочных подстрок, разделенных N-ками или отделенных от спаренного рида пропуском. |
--seedMapMin |
5 |
целое положительное число Минимальная длина затравочных подстрок для начального выравнивания рида. |
--alignIntronMin |
21 |
целое положительное число Минимальный размер интрона: геномный пропуск будет считаться интроном, если его длина больше или равна |
--alignIntronMax |
0 |
целое неотрицательное число Максимальный размер интрона. Если 0, то максимальная дистанция будет определяться по формуле (2ˆ |
--alignMatesGapMax |
0 |
целое неотрицательное число Максимальная дистанция между двумя спаренными ридами. Если 0, максимальная дистанция будет определяться по формуле (
2ˆ |
--alignSJoverhangMin |
5 |
целое положительное число Минимальный оверхенг (размер блока) для сплайсированных выравниваний. |
--alignSJstitchMismatchNmax |
0 -1 0 0 |
четыре целых числа Максимальное число замен для сшивки сплайс-соединений. -1 - неограниченное число. Каждое число обозначает отдельный тип сплайс-сайтов:
|
--alignSJDBoverhangMin |
3 |
целое положительное число Минимальный оверхэнг (размер блока) для выравниваний на аннотированные сплайс-соединения. |
--alignSplicedMateMapLmin |
0 |
целое положительное число Минимальная картируемая длина для сплайсированного спаренного рида. |
--alignSplicedMateMapLminOverLmate |
0.66 |
вещественное положительное число То же, что и |
--alignWindowsPerReadNmax |
10000 |
целое положительное число Максимальное число окон на рид. |
--alignTranscriptsPerWindowNmax |
100 |
целое положительное число Максимальное число транскриптов в окне. |
--alignTranscriptsPerReadNmax |
10000 |
целое положительное число Указывает максимальное число разных выравниваний на рид, по достижении которого поиск новых позиций для выравнивания прекращается. |
--alignEndsType |
Local |
строка Метод выравнивания концевых нуклеотидов рида:
|
--alignEndsProtrude |
0 ConcordantPair |
целое число и строка Допускает ситуацию, когда конец спаренного рида перекрывается с началом рида, но образует выступающий конец (protrusion). В bowtie2 такие пары ридов называются ласточкиным хвостом (dovetail) Например, когда 5’ конец (+) рида оказывается правее, 3’-конца (-) рида. Это возможно когда риды обрезаны с 5’-конца. Параметр принимает число и строку. Число - это максимальное количество выступающих оснований. Строка указывает элайнеру, каким образом записывать пару ридов:
|
--alignSoftClipAtReferenceEnds |
Yes |
строка Позволить ли мягкую обрезку прочтений, пересекающих конец референсной хромосомы.
|
--alignInsertionFlush |
None |
строка Указывает, каким образом переносить позиции вырожденных инсерций (например, когда два нуклеотида АА выравниваются на одну букву А в референсе).
|
Работа со спаренными ридами
| Параметр | Значение по умолчанию | Описание |
|---|---|---|
--peOverlapNbasesMin |
0 |
целое положительное число Минимальное число перекрывающихся оснований, при котором спаренные риды будут соединены и перевыравнены. Значение >0 заставит программу соединять перекрывающиеся спаренные риды. |
--peOverlapMMp |
0.01 |
вещественное неотрицательное число до 1 Максимальная доля ошибочных оснований в области перекрытия ридов. |
Параметры окон для поиска (windows), якорных локусов (anchors) и бинов (bins)
| Параметр | Значение по умолчанию | Описание |
|---|---|---|
--winAnchorMultimapNmax |
50 |
целое положительное число Максимальное число якорных локусов (координаты выравнивания затравочная подстроки, которая имеет минимальное число муножественных выравниваний), на которые нужно проводить выравнивание. |
--winBinNbits |
16 |
целое положительное число логарифм по основанию 2 от размера бинов для окон, в которых будет происходить кластеризация затравочных выравниваний. Каждое окно будет занимать целое число бинов. |
--winAnchorDistNbins |
9 |
целое положительное число Максимальное число бинов между двумя якорными локусами, между которыми возможно объединение якорей в одно окно. |
--winFlankNbins |
4 |
целое положительное число Логарифм по основанию 2 от размера левого или правого франкирующего района в каждом окне. |
--winReadCoverageRelativeMin |
0.5 |
целое неотрицательное число Минимальное относительное покрытие последовательности рида затравочными подстроками в окне. Используется только для алгоритма STARlong. |
--winReadCoverageBasesMin |
0 |
целое положительное число Минимальное число оснований, покрытое затравочной последотельностью в окне. Используется только для алгоритма STARlong. |
Работа с химерными выравниваниями
| Параметр | Значение по умолчанию | Описание |
|---|---|---|
--chimOutType |
Junctions |
строка указывает, каким образом выводить информацию о химерных выравниваниях:
|
--chimSegmentMin |
0 |
целое неотрицательное число Минимальная длина химерных сегментов, если 0, то химерные выравнивания не выводятся. |
--chimScoreMin |
0 |
целое неотрицательное число Минимальная итоговая (после суммирования) оценка для химерных сегментов в выравнивании. |
--chimScoreDropMax |
20 |
целое неотрицательное число Максимальное падение (разница) оценки выравнивания химерных сегментов (сумма оценки за выравнивание для всех химерных сегментов) в сревении с оценкой за целый рид. |
--chimScoreSeparation |
10 |
целое неотрицательное число Минимальная разница (separation) между лучшей оценкой химерного выравнивания и оценкой для другого выравнивания. |
--chimScoreJunctionNonGTAG |
-1 |
целое неотрицательное число Штраф (penalty) за не-GT/AG химерных стыков. |
--chimJunctionOverhangMin |
20 |
целое неотрицательное число Минимальный оверхенг вокруг химерного стыка. |
--chimSegmentReadGapMax |
0 |
целое неотрицательное число Максимальная длина пропуска и последовательности рида между химерными сегментами. |
--chimFilter |
banGenomicN |
строка Разные фильтры для химерных выравниваний:
|
--chimMainSegmentMultNmax |
10 |
целое неотрицательное число Максимальное число множественных вырваниваний для главного химерного сегмента. Если равно 1, то запрещает множественное выравнивание основного химерного сегмента. |
--chimMultimapNmax |
0 |
целое неотрицательное число Максимальное число множественных химерных выравниваний. По умолчанию, разрешены только уникальные химерные выравнивания. |
--chimMultimapScoreRange |
1 |
целое неотрицательное число Устанавливает, на сколько оценка за химерное выравнивание может быть ниже максимального значения, чтобы вывести для данного рида множественные выравнивания. Работает только если задано значение
|
--chimNonchimScoreDropMin |
20 |
целое неотрицательное число Устанавливает величину, определяющую, будет ли программа искать химерные выравнивания. Чем больше, тем реже это будет происходить. Чтобы программа начала искать химерные выравнивания, разница между оценкой за самое лучшее нехимерное выравнивание и оценкой за выравнивание всего рида, должно быть выше заданного числа. |
--chimOutJunctionFormat |
0 |
0 или 1 Указывает на форматирование файла
|
Подсчет числа прочтений в атрибутах генома по аннотации
| Параметр | Значение по умолчанию | Описание |
|---|---|---|
--quantMode |
- |
строка Тип подсчета прочтений в аннотированных районах генома:
|
--quantTranscriptomeBAMcompression |
1 |
целое число от -2 до 10 Уровень сжатия BAM-файла с выравниваниями на транскриптом:
|
--quantTranscriptomeBan |
IndelSoftclipSingleend |
строка
|
Двухкратное картирование ридов для аннотации сайтов сплайсинга
| Параметр | Значение по умолчанию | Описание |
|---|---|---|
--twopassMode |
None |
строка
|
--twopass1readsN |
-1 |
целое число Число ридов, которое нужно выравнить на первом раунде двухкратного выравнивания. Значение |
Параметры WASP
| Параметр | Значение по умолчанию | Описание |
|---|---|---|
--waspOutputMode |
None |
строка Устанавливает параметры вывода из WASP-алгоритма, который используется для аллель-специфичного картирования ридов и поиска молекулярных локусов количественных признаков.
|
Параметры STARsolo (для анализа cingle cell RNA-seq)
| Параметр | Значение по умолчанию | Описание |
|---|---|---|
--soloType |
None |
строка Тип single-cell RNA-seq эксперимента.
|
--soloCBwhitelist |
- |
строка Файл с разрешенным списком (whitelist) баркодов. Только для
|
--soloCBstart |
1 |
целое положительное число Поцизия внутри рида, с которой начинается клеточный баркод. |
--soloCBlen |
16 |
целое положительное число Длина клеточного баркода. |
--soloUMIstart |
17 |
целое положительное число Поцизия внутри рида, с которой начинается клеточный UMI. |
--soloUMIlen |
10 |
целое положительное число Длина UMI. |
--soloBarcodeReadLength |
1 |
целое неотрицательное число Длина баркодированного рида.
|
--soloBarcodeMate |
0 |
целое число Указывает, в каком из спаренных ридов искать баркоды.
|
--soloCBposition |
- |
строка Позиция клеточного баркода в риде с баркодом. Формат строки:
Строки для разных баркодов разделяются пробелами. Например, для inDrop строка будет выглядить следующим образом:
|
--soloUMIposition |
- |
строка То же, что и |
--soloAdapterSequence |
- |
строка Последовательность к якорного баркоду в адаптере. Только одна последовательность разрешена. |
--soloAdapterMismatchesNmax |
1 |
целое неотрицательное число Число разрешенных замен в последовательности адаптера. |
--soloCBmatchWLtype |
1MM_multi |
строка Указывает, каким образом сопоставлять клеточный баркод с белым списком.
|
--soloInputSAMattrBarcodeSeq |
- |
строка Указывает на аттрибуты в SAM-файле, которые будут использоваться для пометки последовательности баркодов. Опция необходима в режиме STARSolo c параметром Например, для 10xСell Ranger или STARsolo BAM-файлов, используются
|
--soloInpu tSAMattrBarcodeQual |
- |
строка Указывает на аттрибуты в SAM-файле, которые будут использоваться для пометки качества баркодов. Опция необходима в режиме STARSolo c параметром Например, для 10xСell Ranger или STARsolo BAM-файлов, используются
По умолчанию параметр имеет значение |
--soloStrand |
Forward |
строка Указывает, есть ли у библиотеке sсRNA-seq специфичность по отношению к цепочке ДНК (read strandness).
|
--soloFeatures |
Gene |
строка Указывает, по каким атрибутам в геноме суммировать UMI. Разные UMI, несущие одинаковый клеточный баркод, будут суммироваться для каждого атрибута:
|
--soloMultiMappers |
-1 |
строка Метод подсчета ридов со множественными выравниваниями. Если 100 ридов выравнивается сразу на 3 гена, то данная опция подскажет, как распределить их между генами.
|
--soloUMIdedup |
1MM_All |
строка Указывает на алгоритм, при помощи которого будет производиться дедупликация ридов. При этом риды с одинаковым UMI будут считаться ПЦР-дупликатами и колапсироваться (будет сохранен только один из всех).
|
--soloUMIfiltering |
- |
строка Метод фильтрации UMI (для ридов, которые картируются на один ген)
|
--soloOutFileNames |
Solo.out/ features.tsv barcodes.tsv matrix.mtx |
путь к папки и имена для файлов названия для файлов вывода STARsolo. Первое слово - префикс к названиям файлов. Это папка, в которую будут сохранены файлы. Второе слово - имя файла с названиями генов. Третье - имя файла для хранения последовательностей баркодов. Последнее слово - имя файла для хранения матрицы с числом прочтений на ген. |
--soloCellFilter |
CellRanger2.2 3000 0.99 10 |
название алгоритма плюс числовые параметры Тип алгоритма фильтрации клеток и параметры.
Подробное описание алгоритма смотреть здесь. Если список параметров не указывать, то это аналогично записи
|
--soloOutFormatFeaturesGeneField3 |
"Gene Expression" |
строка Указывает заголовок для третьего столбика в файле |
--soloCellReadStats |
None |
строка Указывает, выводить ли статистику по ридам для каждого клеточного баркода: |
Источники
Официальная документация к STAR версии 2.7.10b
Оригинальная публикация Dobin 2013