Анализ low-throughput MPRA в стиле STARR-seq
Краткое содержание
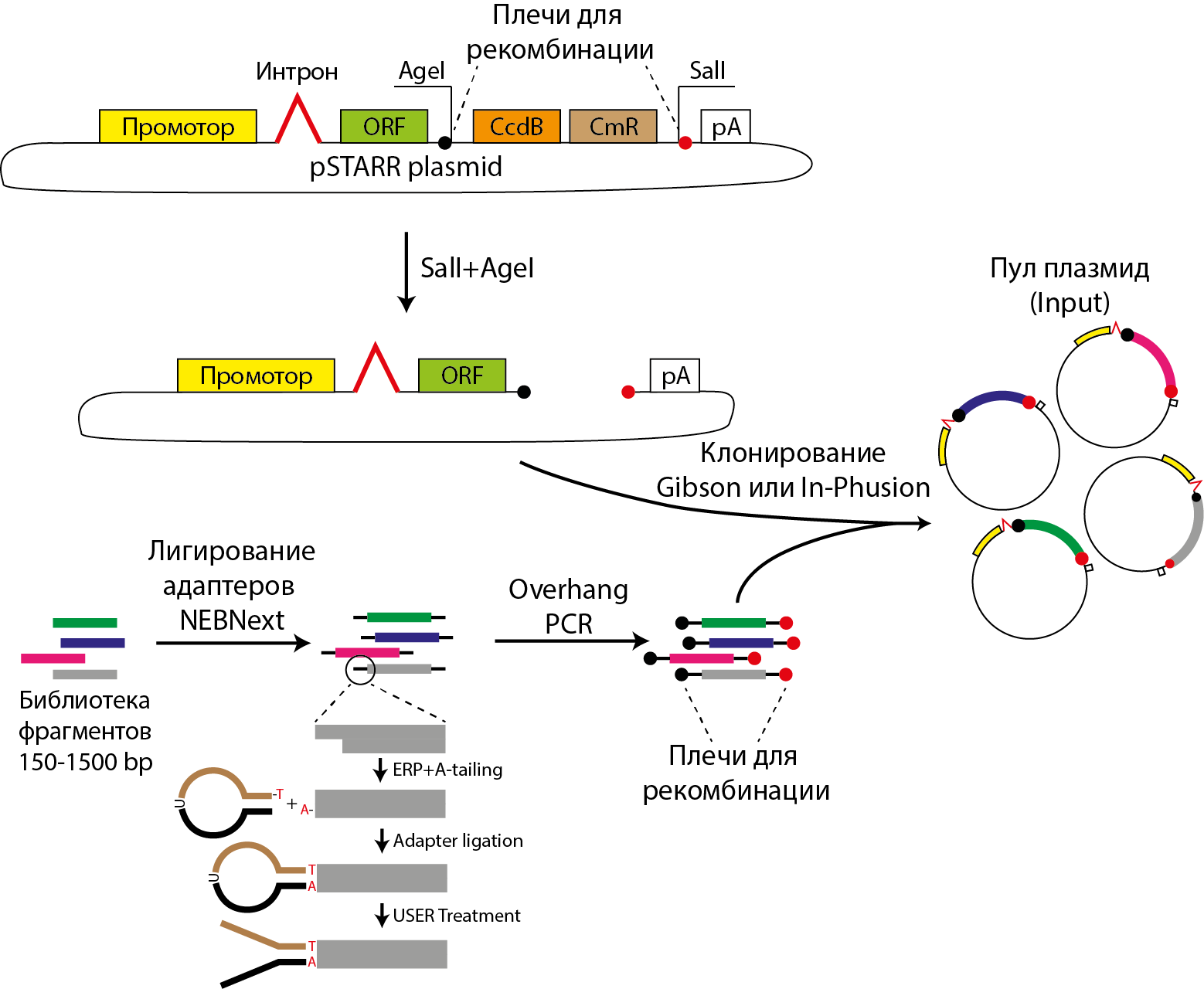
Мы отсеквенировали библиотеки эксперимента STARR-seq в клетках CHO. В ходе эксперимента фрагменты генома CHO-клеток (клетки яичника китайского хомячка), ассоциированные с гистоновой модификацией H3K27ac, были помещены в конструкцию hSTARR-seq_ORI (Addgene #99296). Полученный пул плазмид был трансфецирован в клетки CHO и через два дня из них была выделена РНК. Принципиальная схема эксперимента показана на рисунке ниже.
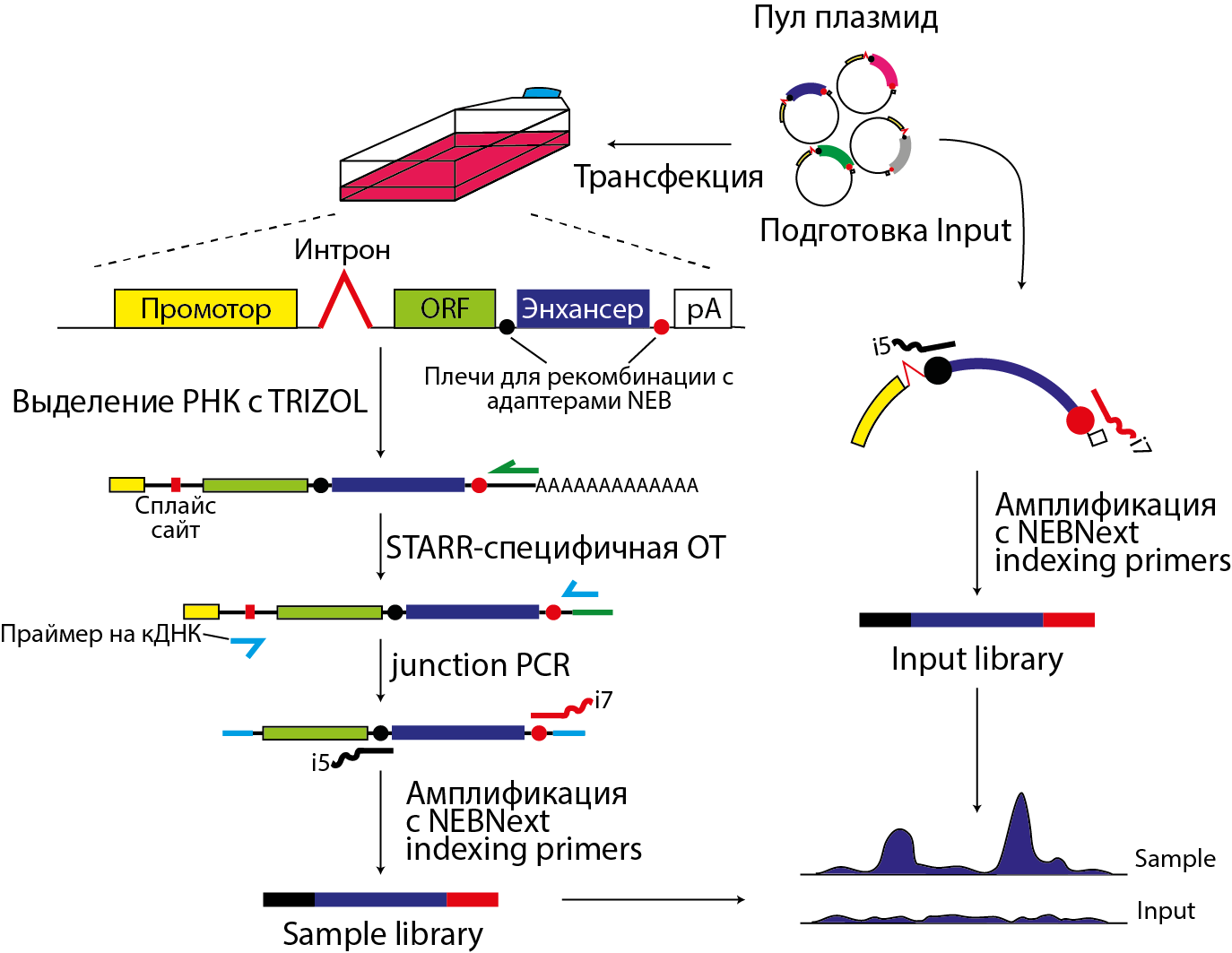
Фрагменты в составе плазмидного пула (Input) и наработанной с плазмид РНК (STARR) были амплифицированы индексированными праймерами NebNEXT в двух технических репликах и отсеквенированы на платформе MiSeq в режиме парных прочтений 2x75bp. Целью показанного ниже биоинформатического анализа являлась идентификация последовательностей, встроенных в плазмидный пул, и оценка их регуляторной активности.
Фрагменты кода и названия файлов представлены с изменениями для более удобного восприятия.
Входные данные
В результате секвенирования имелось 8 файлов в формате fastq.gz. Они были помещены в папку `/home/usr/STARR/`
~$ cd /home/usr/STARR
~/STARR$ ls
Input1_R1.fastq.gz
Input1_R2.fastq.gz
Input2_R1.fastq.gz
Input2_R2.fastq.gz
STARR1_R1.fastq.gz
STARR1_R2.fastq.gz
STARR2_R1.fastq.gz
STARR2_R2.fastq.gz
Парные риды каждой библиотеки помещены в отдельные файлы, помеченные как _R1 и _R2.
Перед анализам желательно глазами просмотреть содержимое файлов. В командной строке Linux это можно сделать следующей командой:
~/STARR$ zcat Input1_R1.fastq.gz | head -n 20
@M02435:118:000000000-JWMPJ:1:1101:13421:1287 1:N:0:1
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
+
###################################
@M02435:118:000000000-JWMPJ:1:1101:14755:1288 1:N:0:1
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
+
###################################
@M02435:118:000000000-JWMPJ:1:1101:13148:1305 1:N:0:1
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
+
###################################
@M02435:118:000000000-JWMPJ:1:1101:12101:1311 1:N:0:1
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
+
###################################
@M02435:118:000000000-JWMPJ:1:1101:14521:1311 1:N:0:1
CTAAGTTTCCCTTTTAACTCATACTTTTCTTTTTTTTCCTTTCTTTTTTTCTTTCTCTTTTTTTTTTCTTTTTTTT
+
6B<,6CFFCCEFGGG,-@CC,E,;CC,6-C<C@,+++,,6,,,,,,<,++,;6,,,,66,;9++7+6,,<,,,+++
Программа zcat считывает содержимое файлов в gzip-архиве и передает в поток стандартного вывода STDOUT (по сути это окно терминала, в котором вы работаете на сервере). Можно перенаправить этот поток к следующей программе при помощи символа | в конце команды вызова zcat. В примере выше перенаправление сделано в программу head, которая выводит только первые n строчек текста в тот же STDOUT. При помощи параметра -n 20 мы заставили head вывести первые 20 строчек.
Вот так выглядят спаренные риды для этой библиотеки:
~/STARR$ zcat Input1_R2.fastq.gz | head -n 20
@M02435:118:000000000-JWMPJ:1:1101:13421:1287 2:N:0:1
CTTTCTTTTTTTCTTCTTTTTTTTTTTTTTTTTTTTTCTCTTTTTTCTCCTTTTCTTTTTTTTTTTTTTTTTTTTT
+
--,86,,-,,++6,;,,;,;,+++++++++7+6+++4,95,,5,,7,,,,,9,5,,99,,++++++++++++++**
@M02435:118:000000000-JWMPJ:1:1101:14755:1288 2:N:0:1
CTATCTTTTTTTCTTCTTTTTTTTTTTTTTTTTATTTCTCTTTTTTCTCCTTTTCTTTTTTTTTTTTTTTTTTTTT
+
--,6,,,-,,++6,;,,;,6,+++++++++7+6,,,969,,,9,,7,,,,,9,9,,95,,++++++++++++++++
@M02435:118:000000000-JWMPJ:1:1101:13148:1305 2:N:0:1
CTTTCTTTTTATCTTCTTTTTTTTTTTTTTTTTTTTTCTCTTTTTTCTCCTTTTCTTTTTTTTTTTTTTTTTTTTT
+
--,6,,,-,,,,-,;,,6,6,+++++++++6+6+++6,66,,6,,44,55,9,9,,55,,+++++++++++++++*
@M02435:118:000000000-JWMPJ:1:1101:12101:1311 2:N:0:1
CTTTCTGTATATCTTCTTTTTTTTTTTTTTTTTATATCTCTTTTTTCTCCTTTTCTTTTTTTTTTTTTTTTTTTTT
+
--,86,,-,,,,6,;,,;,;,+++++++++7+7,,,9,9,,,9,,76,6,,:,96,<9,,++++++++++++++++
@M02435:118:000000000-JWMPJ:1:1101:14521:1311 2:N:0:1
CTTTTTTTCCCCCTTCTTCTTTTCTTCCTTTTCTTCTTCTTTTTCTTTCTCTTTTTCTTTTTCCTTTTTTTCTTTT
+
-8,,,;8@,,,,+7C,,,6;@,;,6;,,,;,,,,6,,,,,,,,,,,,,,;,,;,;,,6,,,66,<,,,,++,,,,5
Легко понять, что на каждое прочтение в файле отведено четыре строки. Первая строка начинается с символа @ и является заголовком, в котором содржится уникальный идентификатор M02435:118:000000000-JWMPJ:1:1101:13421:1287 (у всех ридов они разные) и через пробел служебную последовательность тегов 1:N:0:1. Первое число в теге - это номер рида в паре (1 для _R1 и 2 для _R2). Последнее число в теге - это номер библиотеки. В нашем случае все прочтения из библиотеки Input1 несут теговый номер 1, Input2 - номер 2, STARR1 - номер 3, а STARR2 - номер 4. Это понадобится в дальнейшем.
Обратите внимание, что спаренные прочтения в файлах _R1 и _R2 перечислены в одинаковом порядке. Это видно по идентификаторам. Для программ, которые работают с fastq-файлами это очень важно.
Вторая строка - непосредственно прочтение фрагмента. Третья строка - знак +, который отделяет строку с прочтением от строки с качеством каждого нуклеотида в прочтении. Качество нуклеотида определяется значением Phred-Score, которое может принимать целое значение от 0 (минимальное качество, нуклеотид не опознан) до 41 (максимальное качество, вероятность ошибки ~10-4. Все числа от 0 до 41 закодированы одним из символов, взятых последовательно из таблицы кодировки ASCII (все англоязычные символы на клавиатуре), где числу 0 соответсвует !, а числу 41 соответствует буква I. Такое кодирование Phred-Score называется Phred+33, потому ! является 33-м по счету символом в таблице ASCII. Надо понимать, что нуклеотиды, для которых мы видим символы !, " ,# - это те, что секвенатор не смог распознать. Если же мы види символы английского алфавита, то качество прочтения таких нуклеотидов очень хорошее.
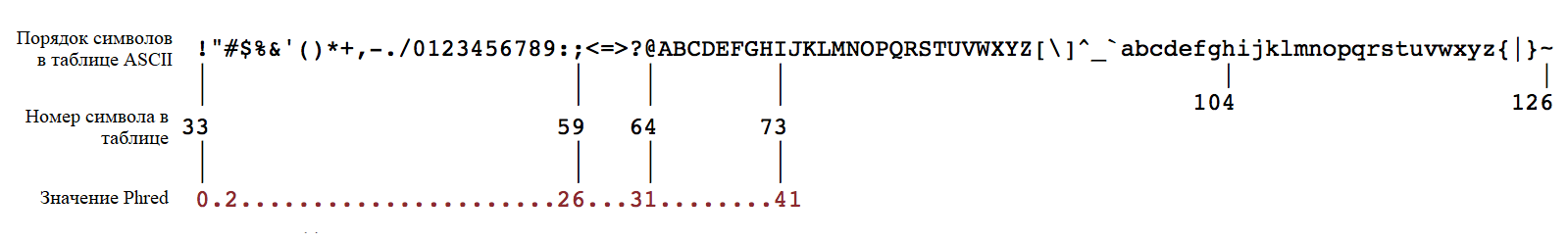
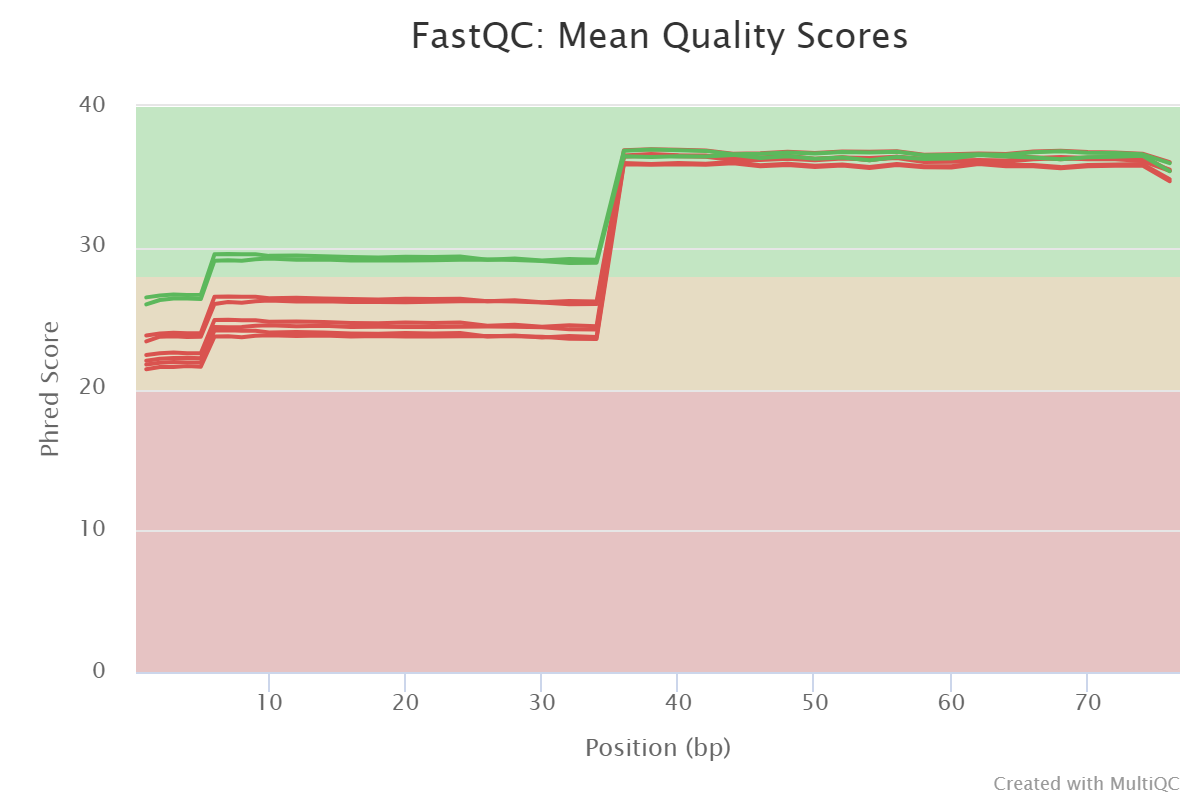
Первое, что бросается в глаза - наличие фрагментов с последовательностью NNNNN… и низким качеством #####.... Такие 36 буквенные риды возникают из-за того, что секвенатор не смог прочитать последовательность, возможно, из-за отсутствия нужного праймера в ней. Быстрый просмотр показывает, что такие неопределенные фрагменты есть во всех образцах и встречатся довольно часто. Это указывает, что во время подготовки библиотек что-то пошло не по плану. Если фрагмент проиндексировался, но чтение с одного из праймеров невозможно, то значит мы имеем дело с праймеровыми димерами (в них отсутсвует одна из последоваетльностей, необхоимых для присоединения затравки на стадии чтения ридов R1 или R2). Это нужно учесть в будущих экспериментах.
Визуальный просмотр прочтений очень полезен, поскольку позволяет выявить недостатки при пробоподготовке библиотек.
Важно отметить, что секвенирование в режиме парных прочтений было выполнено для того, чтобы точно определить фрагменты ДНК с двух сторон, а такие низкокачественные прочтения никак этому не способствуют. Поэтому от них нужно будет избавиться.
Контроль качества
Первым этапом биоинорматического анализа является контроль качества (QC) отсеквенированных библиотек. Ранее мы уже заметили наличие поли-N прочтений длиной 36 букв. Это нужно держать в голове во время интерпретации результатов QC.
Наиболее удобным приложением для QC-оценки fastq-файлов является FastQC. На входе программа принимает файлы в формате .fastq или сжатые fastq.gz, а результаты выдает в виде HTML-отчета, содержащего несколько стандартных диаграмм. На сайте создателей приложения и в большом интернете (например, тут и тут) есть несколько гайдов, объясняющих смысл диаграмм, представленных программой. Ниже будут показаны ключевые пункты, которые были важны для последующего анализа.
Запуск программы FastQC
Программа выдает результат в виде html файла с отчетом и сжатый zip-архив. Название отчета и архива совпадает с название fastq-файла. Так что если делать анализ сразу нескольких fastq-файлов, то в папке попросту будет сложно что-то найти из-за количества файлов. Для удобства советую записывать отчет в отдельную директорию (с помощью параметра -o путь_к_папке). Программа также может работать в мультипоточном режиме, что гораздо быстрее. Число потоков можно указать параметром -t число_потоков. Программа также способна принимать на вход не один файл, а целый список файл1.fastq.gz файл2.fastq.gz файл3.fastq.gz …. Поскольку в нашем случае все fastq-файлы имеют общий фрагмент в названии, то можно обойтись знаком звездочки *.fastq.gz. Интерпретатор командной строки сам сформирует список из файлов, которые имеют одинаковую часть в названии.
~/STARR$ mkdir fastqc
~/STARR$ fastqc -t 50 -o ./fastqc *.fastq.gz
Объединение отчетов fastqc в один файл
Рассматривать каждый отчет fastqc отдельно интересно, но непродуктивно. Чтобы разобраться со всеми библиотеками сразу, можно объединить отчеты в один файл. Это делается при помощи программы MultiQC. На входе она принимает адрес папки, в которой хранятся отчеты FastQC, а на выходе создает единый отчет в формате HTML. Опять же, предлагаю специально для общего отчета создать отдельную папку. Так вы точно его не потеряете среди кучи файлов.
~/STARR$ mkdir multiqc
~/STARR$ multiqc -o multiqc ./fastqc
Предаварительная оценка библиотек
В папке multiqc нас интересует файл с названием multiqc_report.html. В этом отчете есть несколько интерактивных диаграмм. Одна из них показывает общую статистику по библиотекам:
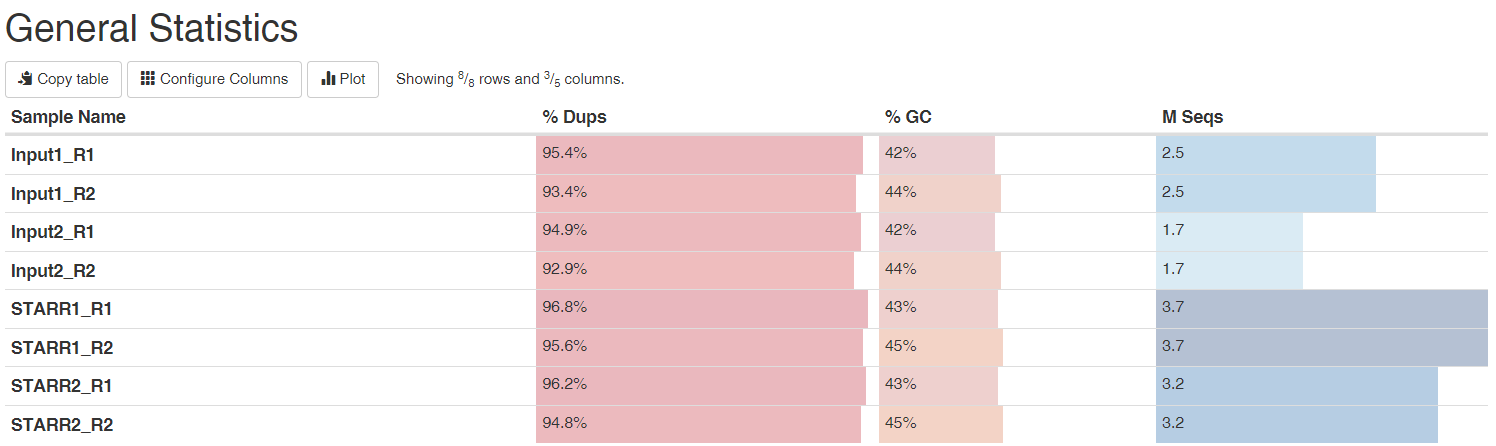
Видно, что до 95% всех фрагментов в библиотеке повторяются. Это означает, что разнообразие библиотек достаточно низкое. Это неудивительно, поскольку цели получить пул плазмид с высоким разнообразием не стояло. Изначально оценка разнообразия пула не далалась (путем подсчета колоний после тарансформации бактерий продуктом реакции клонирования). Но по опыту ожидалось увидеть более тысячи фрагментов. GC-состав в библиотеках приблизительно одинаковый. Это неудивительно, поскольку библиотеки являются техническими репликами. С другой стороны, это косвенно указывает на то, что различия в составе фрагментов при данной глубине секвенирования не велики. Качество прочтения каждого нуклеотида показывает, что качество на 3’-конце прочтений высокое, но в облсти от 1 до 35 оно сильно понижено. Это легко объяснить наличием прочтений NNNNN..., которые мы заметили раньше. Если их удалить, то диаграмма качества, очевидно, станет совершенно нормальной. Это говорит о том, что запуск успешный, проблем с химией быть не должно. Значит, секвенатор показывает нам именно то, что было в библиотеке для секвенирования.
Сколько таких ужасных NNNNN... прочтений в каждой библиотеке? Для ответа на этот вопрос есть распределение числа фрагментов с заданным средним качеством прочтения.
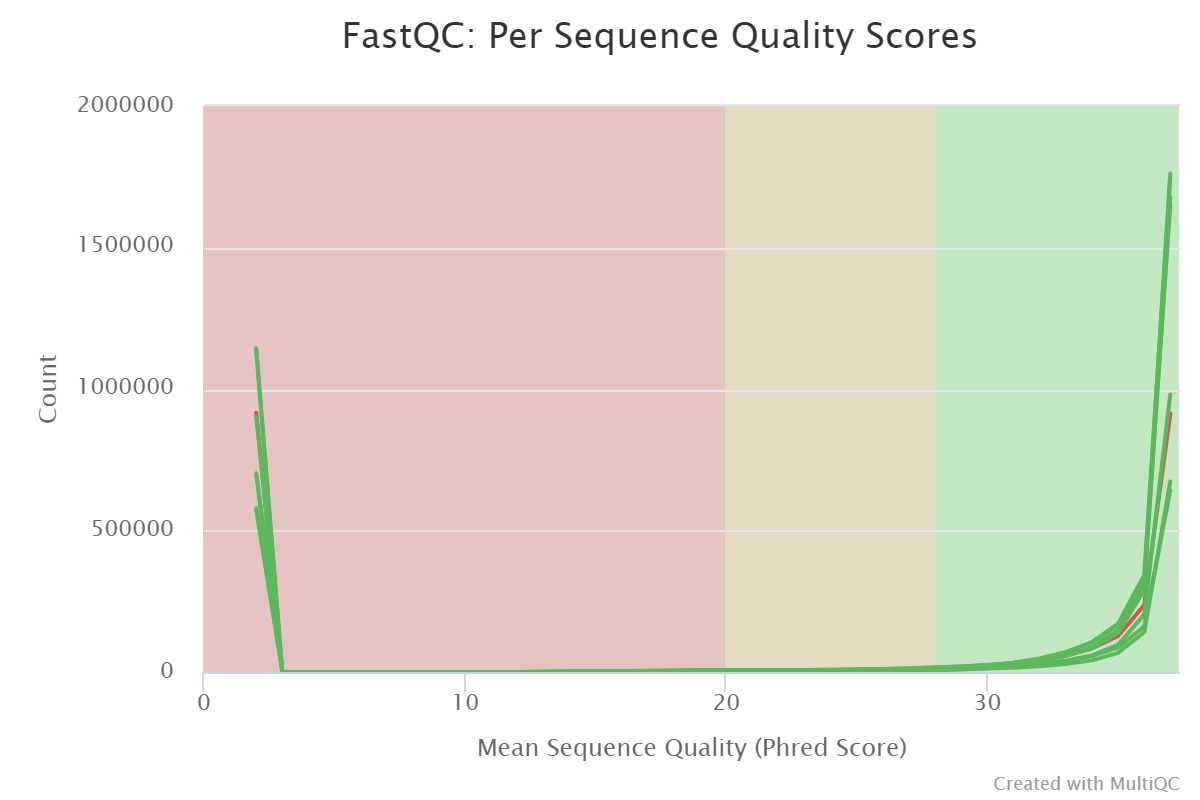
Как видно, не меньше трети всех фрагментов придется удалить из-за проблем с одной из затравочных последовательностей в адаптере. Это указывает на то, что в будущем нужно внимательнее отнестить к этапу обогащения библиотек перед секвенированием.
Мы ожидаем, что разнообразие фрагментов не очень высокое в силу стратегии клонирования плазмидных пулов. Мы также видели, что число дупликатов составляет до 95% от общего числа фрагментов в библиотеках. Предсказать разнообразие фрагментов в пулах можно на основе диаграммы распределения по числу повторов. Здесь показано, что оценочно 35% библиотек представлены последовательностями, повторенными 1-5 тысяч раз, и 35% повторены 10000 раз.
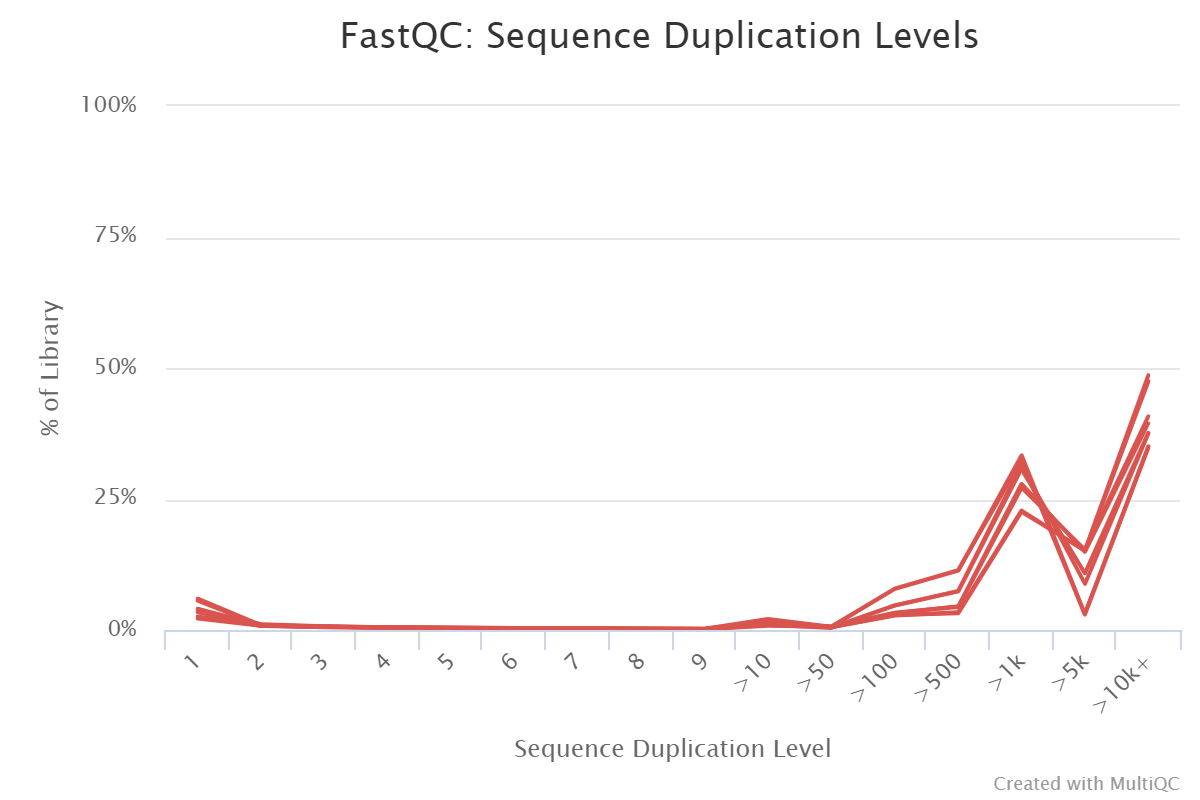
Если число прочтений в библиотеке составляет 1.5 миллиона, треть из этого мы выкинем из-за праймеровых димеров, а в остатке 100 мы увидим сто тысяч уникальных последовательностей, повторенных 1-5 тысяч раз (1.5 миллиона минус треть - миллион. 35% от миллиона - это 250 тысяч, а если это повторы по 1-5 тысяч раз, то в среднем это 100 тысяч). Используя аналогичную прикидку можно прийти к выводу, что в такой библиотеке только 3 десятка уникальных последовательностей будут повторены 10 тысяч раз. Остальные фрагменты повторяются по 1-1000 раз. Таким образом, среди нескольких миллионов прочтений в этих библиотеках мы увидим несколько сотен тысяч уникальных последовательностей ДНК.
Осталось только понять, сколько из уникальных последовательностей возникли из разных участков генома, а не из-за ошибок во время ПЦР на стадии обогащения библиотек секвенирования.
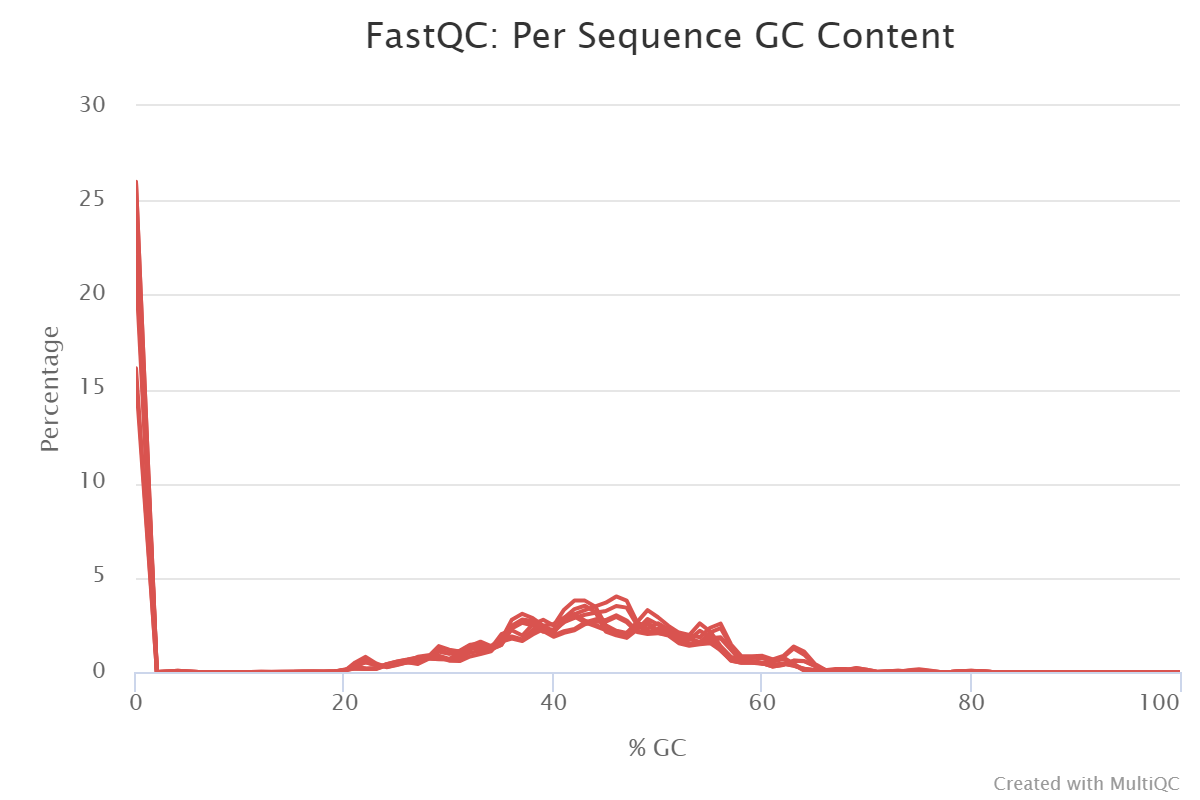
На диаграмме видно, что распределение частоты фрагментов по GC-составу имеет зубчатую форму (на 0 не смотрим, поскольку это прочтения по типу NNNNN.... Это говорит о том, что разнообразие последовательностей недостаточно высокое. Скорее всего, большое число уникальных фрагментов возникли в результате ошибок в ПЦР. В целом, нет ничего удивительного. Мы не должны расстраиваться, поскольку нашей целью не было покрыть геном полностью, плюс протокол, с помощью которого были получены пулы плазмид, не был оптимизирован заранее. Все делалось впервые.
Длина прочтений в запуске была 75 пар. Поскольку фрагменты перед клонированием в плазмиды были получены путем фрагментации генома, то есть вероятность, что часть фрагментов меньше этой длины. В таком случае прочтение не закончится, поскольку вслед за фрагментом идет последовательность адаптера Illumina. Однако если прочтения содержат кусок адаптера, то они не будут выравнены на геном. Нужно ли будет удалять адаптеры? Отчет MultiQC содержит информацию о частоте встречаемости адаптеров Illumina вдоль прочтений:
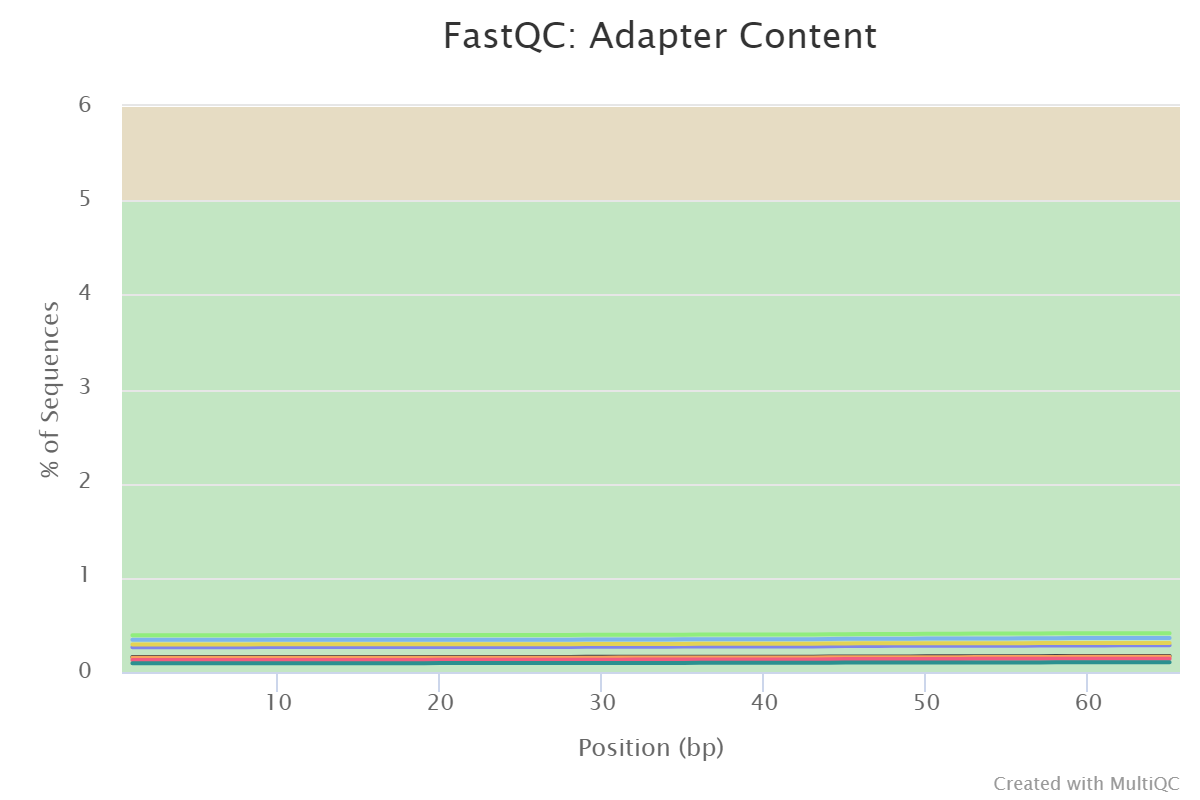
Видно, что есть прочтения, которые начинаются с последовательности адаптера (график в 0 не выходит из 0). Здесь мы имеем дело с димерами адаптеров. В протоколе STARR-seq димеры возникают на стадии лигирования адаптеров во время подготовки фрагментов для клонирования в плазмидный пул. Для нас такие димеры адаптеров могут служить Spike-in контролем, поскольку в таких плазмидах нет регуляторных последовательностей.
Последний график, на который мы обратим внимание - это GC-состав вдоль длины прочтений. Обратите внимание, что в начале и в конце прочтений отсутствует тимин и аденин, соответственно. Это артефакт секвенирования, который повлияет на возможность выравнивания фрагментов на геном. Поэтому все прочтения нужно будет обрезать слева и справа.
Требования к фильтрации прочтений
Для анализа нам теперь нужно принять решение, каким образом избавляться от мусора в сиквенсах. Ранее упоминалось, что нужно сделать следующее:
- Удалить фрагменты, в которых хотя бы одно парное прочтение не удалось (те самые
NNNNN...) - Удалить адаптеры
- Обрезать прочтения слева и справа на 1-2 буквы
- Отфильтровать итоговые укороченные прочтения по длине так, чтобы их можно было качественно выравнить на геном
- Отфильтровать димеры адаптеров, чтобы использовать как Spike-in
Благодаря внимательному QC мы не только определили качество запуска, но и оценили разнообразие фрагментов в плазмидных пулах (несколько тысяч), а также сформулировали рекомендации для дальнейшего улучшения лабораторного протокола STARR-seq.
Схема анализа
Алгоритм анализа схематически представлен на следующем рисунке:
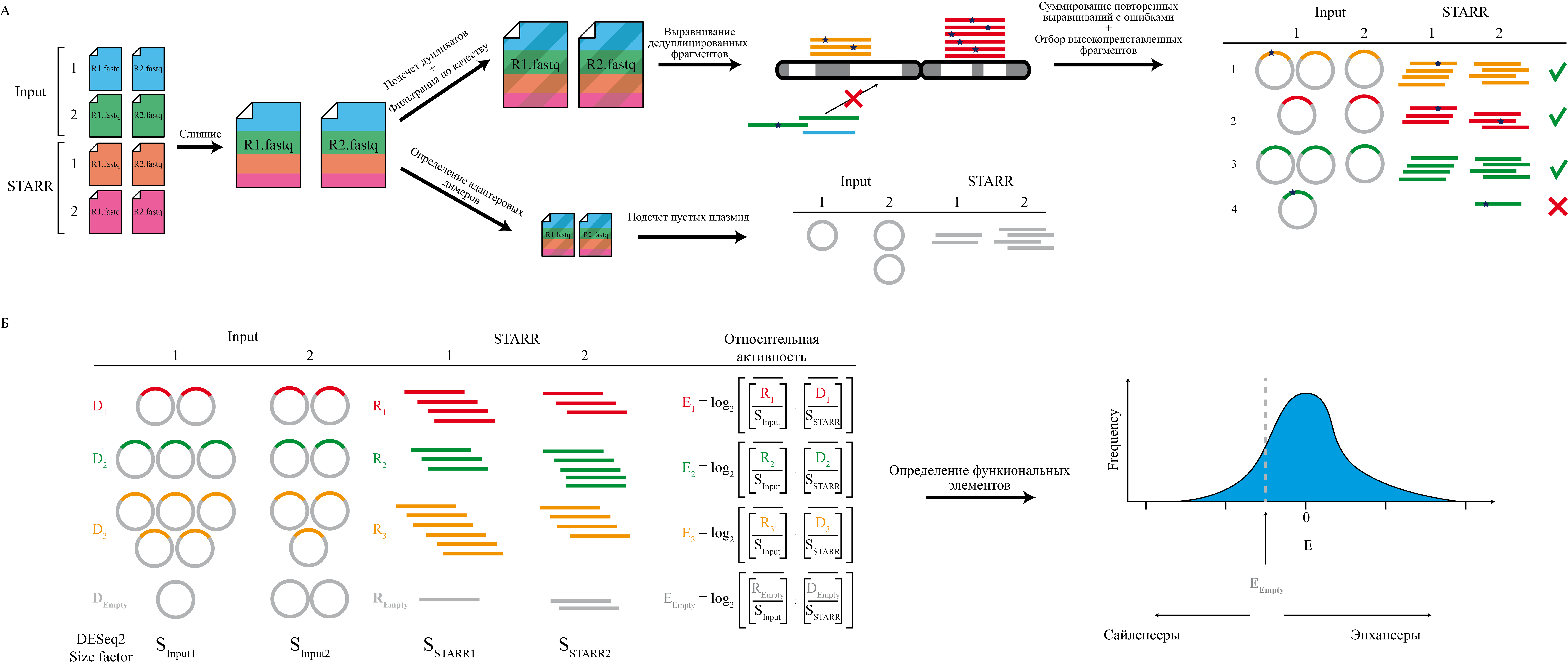
На схеме А показано, что анализ включает несколько этапов. На первом этапе мы объединим все прочтения, сделанные с одной стороны, в один файл. Это удобно, поскольку заголовки прочтений несут тег, соответствующий типу библиотеки. Далее мы отфильтруем риды двумя способами:
- димеры адаптеров отфильтруем в один файл;
- остальные прочтения обрежем и отфильтруем, как было сказано выше.
С помощью дедупликации мы идентифицируем в вдусторонних прочтениях идентичные фрагменты ДНК. Если идентичные фрагменты в процессе подготовки библиотек для секвенирования накопили ошибки, то алгоритм дедупликации не опознает их как копии. Однако мы можем выравнить фрагменты на геном CHO-клеток и объединить те фрагменты, которые выравниваются в одно и то же место генома. Мы также можем использовать фильтрацию по количеству, чтобы удалить фрагменты с низкой представленностью. Прошедшие фильтрацию уникальные последовательности с большой вероятностью не являются продуктом ошибочной амплификации.
На схеме Б показана схема анализа активности фрагментов, выявленных на прошлом этапе. Зная представленность каждого фрагмента в отсеквенированных библиотеках, мы можем использовать в качестве меры активностим величину E=log2FoldChange - показатель, характеризующий изменение представленности фрагмента в пуле отсеквенированных фрагментов.
Для подсчета этой величины мы делим представленность каждого фрагмента в данной библиотеке на коэффициент, пропорциональный общему количеству фрагментов в библиотеке. Этот коэффициент, size factor, вычисляется программой DESeq2 при помощи алгоритма медианы отношений (см. сюда). Почему именно size factor? На самом деле нам без разницы, мы могли бы использовать сумму по всем фрагментам в библиотеке, однако этот коэффициент менее чувствителен к ситуации, когда какой-то из фрагментов перепредставлен в одной библиотеке из четырех. То есть size factor лучше подходит для сравнения разных образцов. После нормировки при помощи size factor мы находим среднее значение для отношения между репликами Input и STARR, а затем находим FoldChange - отношение нормированных величин в пулах мРНК по сравнению с пулом плазмид. После логарифмирование получается число, которое нормально распределено относительно нуля.
Если в результате транскрипции представленность фрагмента увеличилась по сравнению с другими фрагментами (log2FoldChange>0), то мы можем с надежностью утверждать, что данный фрагмент ДНК увеличивает транскрипцию сильнее, чем остальные фрагменты. Однако мы все еще не знаем, являются ли такие фрагменты энхансерами “по определению”, ведь совершенно неясно, какой log2FoldChange будут демонстрировать последовательности, которые не влияют на транскрипцию. Именно по этой причине мы извлекаем из данных количество “пустых плазмид”, которые на сиквенсах выглядят как димеры адаптеров. По умолчанию мы считаем такие плазмиды не имеющими регуляторных последовательностей, а значит их уровень log2FoldChange можно считать базовым для определения энхансерной активности.
Объединение прочтений в один файл
Как упоминалось выше, заголовки перед каждым прочтением в fastq-файлах содержат последовательнось тегов, по которым можно идентифицировать пару, а также образец. Благодаря этому мы можем спокойно объединить образцы, чтобы в последующем не записивыть одну и ту же команду несколько раз.
Ниже мы исползовали программу zcat, которая считывает текстовые файлы из gzip-архивов и построчно передает в STDIN (в окно терминала). На вход программа принимает список из fastq.gz файлов. Далее мы приказываем программе записать все строки в fastq-файл с названием merged_R1.fastq или merged_R2.fastq (zcat не будет сжимать файл вывода). После этого мы воспользовались программой архиватором gzip, которая сжала эти файлы и добавила к названию суффикс .gz, а затем вывели названия всех файлов, содержащих слово “merged” в названии, чтобы убедиться в этом.
~/STARR$ zcat Input1_R1.fastq.gz Input2_R1.fastq.gz STARR1_R1.fastq.gz STARR2_R1.fastq.gz > merged_R1.fastq
~/STARR$ gzip merged_R1.fastq
~/STARR$ zcat Input1_R2.fastq.gz Input2_R2.fastq.gz STARR1_R2.fastq.gz STARR2_R2.fastq.gz > merged_R2.fastq
~/STARR$ gzip merged_R2.fastq
~/STARR$ ls merged*
merged_R1.fastq.gz
merged_R2.fastq.gz
Важно, что в полученных файлах merged_R1.fastq.gz и merged_R2.fastq.gz спаренные риды перечисляются в одинаковом порядке.
Фильтрация адаптеровых димеров
Найти димеры адаптеров можно при помощи Cutadapt. Мы запустим программу в два этапа. На первом мы удалим все пары ридов, в которых есть NNNNN... хотя бы в одном из ридов. Это делается при помощи параметров --max-n 5 --pair-filter=any. Здесь же мы установим последовательности для поиска адаптеров, которые можно найти на сайте Illumina в разделе Adapter Trimming. Обратите внимание, что адаптеры в прочтениях R1 и R2 отличаются, но на 5’-конце несут одинаковую последовательность длиной 13 букв:
| Read 1 | AGATCGGAAGAGCACACGTCTGAACTCCAGTCA |
| Read 2 | AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT |
Последовательность адаптера, которую нужно будет обрезать в первом риде указываются после параметра -a. Также есть возможность указать возможное количество max_error_rate=0.2, и минимальное перекрытие адаптера с прочтением min_overlap=5. Эти дополнительные параметры указываются вслед за последовательностью через точку с запятой и в кавычках:
-a "AGATCGGAAGAGCACACGTCTGAACTCCAGTCA;max_error_rate=0.2;min_overlap=5"
Аналогично, пишем для прочтения R2, только с использованием параметра -A:
-A "AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT;max_error_rate=0.2;min_overlap=5"
Нас интересуют только прочтения, в каждом из которых в каждой паре нашлись адаптеры. Чтобы заставить отобрать такие прочтения мы воспользуемся параметром --discard-untrimmed.
После того, как такие риды будут найдены, нам нужно будет снова воспользоваться Cutadapt’ом, чтобы отобрать те прочтения, в которых после удаления адаптера длина стала минимальной. Мы возьмем не 0, а
~/STARR$ cutadapt --max-n 5 --pair-filter=any -j 60 -a "AGATCGGAAGAGCACACGTCTGAACTCCAGTCA;max_error_rate=0.2;min_overlap=5" -A "AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT;max_error_rate=0.2;min_overlap=5" --discard-untrimmed --interleaved merged_R1.fastq.gz merged_R2.fastq.gz | cutadapt -j 60 --maximum-length=3 --interleaved --pair-filter=any -o merged_empty_R1_filt.fastq.gz -p merged_empty_R2_filt.fastq.gz -